

发布者:Primerbank 时间:2017-09-22 浏览量:1372
· Ruslan Kalendar , David Lee , Alan H. Schulman
The polymerase chain reaction is fundamental to molecular biology and is the most important practical molecular technique for the research laboratory. We have developed and tested efficient tools for PCR primer and probe design, which also predict oligonucleotide properties based on experimental studies of PCR efficiency. The tools provide comprehensive facilities for designing primers for most PCR applications and their combinations, including standard, multiplex, long-distance, inverse, real-time, unique, group-specific, bisulphite modification assays, Overlap-Extension PCR Multi-Fragment Assembly, as well as a programme to design oligonucleotide sets for long sequence assembly by ligase chain reaction. The in silico PCR primer or probe search includes comprehensive analyses of individual primers and primer pairs. It calculates the melting temperature for standard and degenerate oligonucleotides including LNA and other modifications, provides analyses for a set of primers with prediction of oligonucleotide properties, dimer and G-quadruplex detection, linguistic complexity, and provides a dilution and resuspension calculator.
PCR primer design; Primer linguistic complexity; Sequence assembly; Software; Probe design; Ligase chain reaction
The polymerase chain reaction (PCR) is fundamental to molecular biology and is the most important practical molecular technique for the research laboratory. However, the utility of the method is dependent on identifying unique primer sequences and designing PCR-efficient primers. Primer design is a critical step in all types of PCR methods to ensure specific and efficient amplification of a target sequence [1]; [2]; [3]; [4]; [5]; [6]; [7]; [8]; [9] ; [10]. Even though there are currently many online and commercial bioinformatics tools, primer design for PCR is still not as convenient and practical as it might be for routine use. The adaptation of PCR for different applications has made it necessary to develop new criteria for PCR primer and probe design to cover uses such as RT-PCR, qPCR, group-specific [2]; [6]; [7]; [8] ; [11] unique PCR, combinations of multiple primers in multiplex PCR, discovery of simple sequence repeats and their amplification as diagnostic markers, IRAP/REMAP [12], TaqMan, and molecular beacon and microarray oligonucleotides.
In developing Java web tools (Table 1), our aim was to create practical and easy-to-use software for routine manipulation and analysis of sequences for most PCR applications. The parameters adopted are based on our experimental data for efficient PCR and are translated into algorithms in order to design combinations of primer pairs for optimal amplification. The Java web tools are based on our FastPCR software for Windows [2] and together have been successfully used throughout the scientific community in a wide range of PCR-related applications.
Table 1.
Summary of Java web tools for PCR, in silico PCR, oligonucleotide assembly and analysis.
|
Features |
|
PCR tool provides comprehensive facilities for designing primers for most PCR applications and their combinations: |
|
standard, multiplex, long distance, inverse, real-time, unique or group-specific, LATE-PCR, bisulphite modification assays, polymerase extension PCR multi-fragment assembly cloning; |
|
in silico (virtual) PCR or multiple primer or probe searches, prediction of probable PCR products, and search for potential mismatching locations of the specified primers or probes; |
|
design of oligonucleotide sets for long sequence assembly by ligase chain reaction (LCR) and PCR; |
|
testing of individual primers, melting temperature calculation for standard and degenerate oligonucleotides including LNA and other modifications; |
|
PCR efficiency, linguistic complexity, dimer and G-quadruplex detection, dilution and resuspension calculator; |
|
analysis of features of multiple primers simultaneously, including Tm, GC content, linguistic complexity, dimer formation; optimal Ta; |
|
identification of simple sequence repeat (SSR) loci; |
|
pattern analysis of sequences including (G − C)/(G + C), (A − T)/(A + T), (S − W)/(S + W) and purine–pyrimidine (R − Y)/(R + Y) skews, CG% content, primer quality, Tm, and linguistic sequence complexity |
|
generation of pipetting tables for setting up PCR or qPCR reactions. |
Primer design is one of the key steps for successful PCR. For PCR applications, primers are usually 18–35 bases in length and should be designed such that they have complete sequence identity to the desired target fragment to be amplified. The parameters, controllable either by the user or automatically, are primer length (12–500 nt), melting temperature for short primers calculated by nearest neighbour thermodynamic parameters, the theoretical primer PCR efficiency (quality %) value, primer CG content, 3′ end terminal enforcement, preferable 3′ terminal nucleotide sequence composition in degenerated formulae, and added sequence tags at 5′ termini. The other main parameters used for primer selection are: the general nucleotide structure of the primer such as linguistic complexity (nucleotide arrangement and composition); specificity; the melting temperature of the whole primer and the melting temperature at the 3′ and 5′ termini; self-complementarity; secondary (non-specific) binding.
The software can dynamically optimise the best primer length for entered parameters. All PCR primer (probe) design parameters are flexible and changeable according to the specifics of the analysed sequence and task. Primer pairs are analysed for cross-hybridisation, specificity of both primers and, optionally, selected with similar melting temperatures. Primers with balanced melting temperatures (within 1–6 °C of each other) are desirable but not mandatory. The default primer design selection criteria are shown in Table 2. It is possible to use pre-designed primers or probes or, alternatively, pre-designed primers can act as references for the design of new primers. The programme accepts a list of pre-designed oligonucleotide sequences and checks the compatibility of each primer with a newly designed primer or probe.
Table 2.
Default primer design selection criteria.
|
Criteria |
Default |
Ideal |
|
Length (nt) |
19–21 |
> 21 |
|
Tm range (°C)a |
52–68 |
60–68 |
|
Tma 12 bases at 3′-end |
34–48 |
41–47 |
|
GC (%) |
45–65 |
50 |
|
3′-end composition (5′-nnn-3′) |
sww, sws, ssw, wss |
ssa, sws, wss |
|
Sequence linguistic complexity (LC,%) b |
> 80 |
> 95 |
|
Sequence Quality (PQ,%) |
> 80 |
> 95 |
a Nearest neighbour thermodynamic parameters (11).
b Sequence linguistic complexity measurement was performed using the alphabet-capacity L-gram method.
The programme is able to generate either long oligomers or PCR primers for the amplification of gene-specific DNA fragments of user-defined length. Up to now, several publicly available primer/oligo design programmes have been developed [3]; [4]; [5]; [6]; [7]; [8] ; [11]. All of them are specialised for either the design of PCR primers or oligomers; some of them are based on the Primer3 code [3]. jPCR is based on our unique FastPCR software and its fast and efficient code; it provides a more flexible approach to designing primers for many applications. It will check if either primers or probes have secondary binding sites in the input sequences that may give rise to an additional PCR product. The selection of the optimal target region for the design of long oligomers is performed in the same way as for PCR primers. The basic parameters in primer design are also used as a measure of the oligomer quality and the thermodynamic stability of the 3′ and 5′ terminal bases are evaluated.
The proposal of primer pairs and the selection of the best pairs are possible. The user can vary the product size or design primer pairs for the whole sequence without specifying parameters by using default or pre-designed parameters. The pre-designed parameters are specified for different situations: for example, for sequences with low CG content or long distance PCR, or degenerated sequences, or for manual input. Results show the list of best primer candidates and all compatible primer pairs that are optimal for PCR. Users can specify, individually for each sequence, multiple locations for both forward and reverse primer designs inside each sequence, whilst PCR design will be performed independently for different targets. Multiplex PCRs can be performed simultaneously within a single sequence with multiple amplicons as well as for different sequences, or combinations of both. The user can specify the PCR product size in a similar way.
The Tm is defined as the temperature at which half the DNA strands are in the double-helical state and half are in the “random-coil” state. The Tm for short oligonucleotides with normal or degenerate (mixed or “wobble”) nucleotide combinations is calculated in the default setting using nearest neighbour thermodynamic parameters [13]; [14]; [15]; [16] ; [17]. The CG content of an oligonucleotide is the most important factor that influences the Tm value. The melting temperature for mixed bases is calculated by averaging nearest neighbour thermodynamic parameters – enthalpy and entropy values – at each mixed site; extinction coefficient is similarly predicted by averaging nearest neighbour values at mixed sites [5]. Table S1 shows the nearest neighbour thermodynamic parameters, the enthalpy and entropy values for normal and mixed nucleotides. The first nucleotide in 5′N1N2 are shown in the horizontal column and the second nucleotide 5′N1N2 in the vertical column.
The sequence complexity calculation method can be used to search for conserved regions between compared sequences for the detection of low-complexity regions including simple sequence repeats, imperfect direct or inverted repeats, polypurine and polypyrimidine triple-stranded DNA structures, and four-stranded structures (such as G-quadruplexes). Linguistic complexity (LC) measurements are performed using the alphabet-capacity L-gram method [18] ; [19] along the whole sequence length and calculated as the sum of the observed range (xi) from 1 to L size words in the sequence divided by the sum of the expected (E) value for this sequence length. G-rich (and C-rich) nucleic acid sequences can fold into four-stranded DNA structures that contain stacks of G-quartets [see more at http://www.quadruplex.org/]. These quadruplexes can be formed by the intermolecular association of two or four DNA molecules, dimerisation of sequences that contain two G-bases, or by the intermolecular folding of a single strand containing four blocks of guanines [20]; [21]; [22]; [23]; [24]; [25] ; [26]; these are easy to eliminate from primer design because of their low linguistic complexity, LC = 32% for (TTAGGG)4.
The programme includes various bioinformatics tools for pattern analysis in sequences having GC skew, (G − C)/(G + C), AT skew, (A − T)/(A + T), CG − AT skew, (S − W)/(S + W), or purine–pyrimidine (R − Y)/(R + Y) skew regarding CG content and melting temperature and considers linguistic sequence complexity profiles. For example the GC skew in a sliding window of n bases is calculated with a step of one base, according to the formula, (G − C)/(G + C), in which G is the total number of guanines and C is the total number of cytosines for all sequences in the windows [27]. Positive GC-skew values indicated an overabundance of G bases, whereas negative GC-skew values represented an overabundance of C bases. Similarly, other skews are calculated in the sequence.
Our experimental data showed that the primer nucleotide composition and melting temperature of the 12 bases at the 3′ end of the primers are important factors for PCR efficiency. The melting temperature of the 12 base 3′ terminus is calculated preferably by nearest-neighbour thermodynamic parameters [15]. The composition of the sequence at the 3′ terminus is important; primers with two terminal C/G bases are recommended for increased PCR efficiency [28]; [29] ; [30]. Nucleotide residues C and G form a strong pairing structure in the duplex DNA strands. Stability at the 3′ end in primer template complexes will improve the polymerization efficiency.
We specify an abstract parameter called Primer Quality (PQ) that can help to estimate the efficiency of primers for PCR. PQ is calculated by the consecutive summation of the points according to the following parameters: total sequence and purine–pyrimidine sequence complexity, the melting temperatures of the whole primer and of the terminal 3′ and 5′ 12 bases. Self-complementarity, which gives rise to possible dimer and hairpin structures, reduces the final value. PQ tries to describe the likelihood of PCR success of each primer; this value varies from 100 for the best to 0 for the worst primers (Fig. 1).
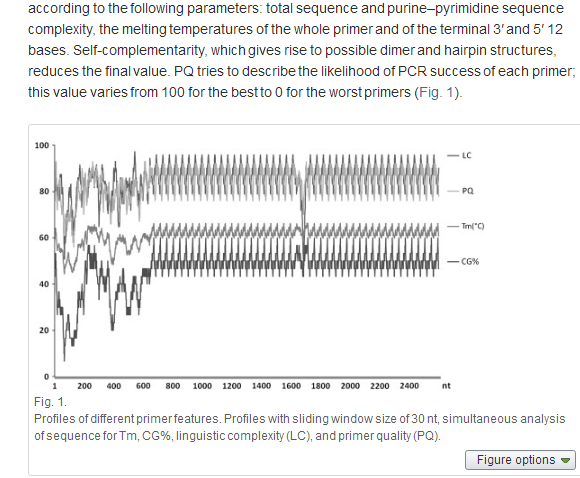
To meet multiplexing demands, it is possible in the programme to select the best primer with an optimal temperature range, allowing the design of qualified primers or probes for any target sequence with any CG and repeat content. PQ values of 80 and higher allow for the rapid choice of the best PCR primer pair combination. No adverse effects, due to the modification of the reaction buffer, chosen thermostable polymerases, or variations in annealing temperature, have been observed on the reproducibility of PCR amplification using primers with high PQ.
Primer-dimers involving one or two sequences may occur in a PCR reaction. The jPCR tool eliminates intra- and inter-oligonucleotide reactions before generating a primer list and primer pair candidates. It is very important for PCR efficiency that the production of stable and inhibitory dimers is avoided, especially avoiding complementarity in the 3′-ends of primers from whence the polymerase will extend. Stable primer dimer formation is very effective at inhibiting PCR since the dimers formed are amplified efficiently and compete with the intended target (Fig. 2).

Primer dimer prediction is based on analysis of non-gap local alignment and the stability of both the 3′ end and the central part of the primers. Primers will be rejected when they have the potential to form stable dimers with at least 5 bases at the 3′ end or 7 bases at the central part. Tools calculate Tm for primer dimers with mismatches for pure, mixed, or modified (inosine, uridine, or locked nucleic acid) bases using averaged nearest neighbour thermodynamic parameters provided for DNA/DNA duplexes [13]; [14]; [15]; [16]; [31] ; [32]. Besides Watson–Crick base pairing, there is a variety of other hydrogen bonding configurations possible [20]; [21]; [22]; [23]; [24]; [25] ; [26] such as G-quadruplexes (Fig. 3). By default, the software predicts the presence of putative G-quadruplexes in primer sequences.

The optimal annealing temperature (Ta) is the range of temperatures where efficiency of PCR amplification is maximal without non-specific products. The most important values for estimating the Ta is the primer quality, the Tm of the primers and the length of PCR fragment. Primers with high Tm's (> 60 °C) can be used in PCRs with a wide Ta range compared to primers with low Tm's (< 50 °C). The optimal annealing temperature for PCR is calculated directly as the value for the primer with the lowest Tm (Tmmin). However, PCR can work in temperatures up to 10 °C higher than the Tm of the primer, especially when reactions contain high primer concentrations (0.6–1.0 μM) to favour primer target duplex formation:
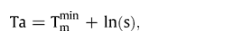
where s is length of PCR fragment.
In our experience and of those of the more than 400 users of FastPCR on which the jPCR algorithms are based (http://www.primerdigital.com/fastpcr/citations.html), almost all high-quality primers designed by jPCR in the default or “best” mode provide amplification at annealing temperatures from 60 to 72 °C without loss of PCR efficiency, and show good amplification in varying PCR annealing temperatures and when using different DNA polymerases and buffers. Comparisons of jPCR to popular tools for PCR primer design and oligonucleotide analysis including the most popular on-line primer design and analysis packages have been posted http://primerdigital.com/tools/soft.html.
Individual and sets of primers are evaluated using jPCR, PrimerAnalyser or PrimersList software. They calculate primer Tm's using default or other formulae for normal and degenerate nucleotide combinations, CG content, extinction coefficient, unit conversion (nmol per OD), mass (μg per OD), molecular weight, linguistic complexity, and primer PCR efficiency. Users can select either DNA or RNA primers (PrimerAnalyser) with normal or degenerate oligonucleotides or modifications with different labels (for example inosine, uridine, or fluorescent dyes). Tools allow the choice of other nearest-neighbour thermodynamic parameters or non-thermodynamic Tm calculation formulae. For example, for non-thermodynamic Tm calculation of oligonucleotides, we suggest using the simple formulae:
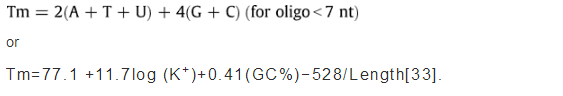
For locked nucleic acid (LNA) modifications the four symbols: dA = E, dC = F, dG = J, dT = L are used. Both programmes perform analyses on-type, which allow users to see the results immediately on screen. They can also calculate the volume of solvent required to attain a specific concentration from the known mass (mg), OD, or moles of dry oligonucleotide.
All primers are analysed for intra- and inter-primer interactions to form dimers. Primer(s) can efficiently hybridise using the 5′ end or middle of the sequences. Even though such interactions are not efficiently extended by DNA polymerase, their formation, however, reduces the effective primer concentration available for binding to the target and their presence can strongly inhibit PCR since double-stranded DNA at high concentrations is a strong inhibitor of DNA polymerase.
The specificity of the oligonucleotides is one of the most important factors for good PCR; optimal primers should hybridise only to the target sequence, particularly when complex genomic DNA is used as the template. Amplification problems can arise due to primers annealing to repetitious sequences (retrotransposons, DNA transposons, or tandem repeats). Alternative product amplification can also occur when primers are complementary to inverted repeats and produce multiple bands. This is unlikely when primers have been designed using specific DNA sequences (unique PCR). However, the generation of inverted repeat sequences is exploited in two common generic DNA fingerprinting methods — RAPD and AP-PCR [34] ; [35]. Because only one primer is used in these PCR reactions, the ends of the products must be reverse complements and thus can form stem-loops.
The techniques of inter-retrotransposon amplification polymorphism (IRAP), retrotransposon-microsatellite amplification polymorphisms (REMAP), inter-MITE amplification [12]; [36] ; [37], and Alu-repeat polymorphism [38] ; [39] have exploited these highly abundant dispersed repeats as markers. However, primers complementary to repetitious DNA may produce many non-specific bands in single-primer amplification and compromise the performance of unique PCRs. A homology search of the primer sequence, for example using ‘blastn’ against all sequences in GenBank or EMBL-Bank, will determine whether the primer is likely to interact with dispersed repeats. Alternatively, one can create a small local specialised library of repeat sequences based on those in Repbase [40] or TREP [http://wheat.pw.usda.gov/ITMI/Repeats/].
By default, jPCR performs a non-specific binding test for each given sequence. Additionally the software allows this test to be performed against a reference sequence or sequences (e.g. BAC, YAC) or one's own database. Primers that bind to more than one location on current sequences will be rejected. Even though the non-specific primer binding test is performed as a default for all primers, the user may cancel the operation. Identification of secondary binding sites including mismatched hybridisation is normally performed by considering the similarity of the primer to targets along the entire primer sequence. An implicit assumption is that stable hybridisation of a primer with the template is a prerequisite for priming by DNA polymerase. The jPCR pays particular attention to the 3′ end portion of the primer and calculates the similarity of 3′ end of the primer to target (the length is chosen by the user) to determine the stability of the 3′-terminus. The secondary non-specific primer binding test is based on a quick, non-gapped local alignment (that allows one mismatch within a hash index of 9-mers) screening between the reference and input sequence.
Multiplex PCR is an approach commonly used to amplify several DNA target regions in a single reaction. The simultaneous amplification of many targets reduces the number of reactions that needs to be performed; multiplex PCR thus increases throughput efficiency. The design of multiplex PCR assays can be difficult because it involves extensive computational analyses of primer pairs for cross interactions. To achieve uniform amplification of the targets, the primers must be designed to bind with equal efficiencies to their targets. jPCR can quickly design a set of multiplex PCR primers for all the input sequences and/or multiplex targets within each sequence. PCR conditions may need to be adjusted; for example, the annealing temperature increased or lowered so that all products are amplified equally efficiently. To achieve this, most existing multiplex primer design packages use primer melting temperature.
In practical terms, the design of almost identical Ta's and Tm's is very important. The melting temperatures of the PCR products are also important since these are related to annealing temperature values. The Tm of a PCR product depends on its GC content and length; short products are more efficiently amplified at low PCR annealing temperatures (100 bp, 55 °C) than long products (3000 bp, > 60 °C). For most multiplex PCRs, there is usually a small variation (up 5 °C) between the optimal Ta's of all primer pairs and PCR products. The annealing temperature must be optimal in order to maximise the likelihood of amplifying the target genomic sequences whilst minimising the risk of non-specific amplification. Further improvements can be achieved by selecting the optimal set of primers that maximise the range of common Tm's. Once prompted, jPCR calculates multiplex PCR primer pairs for given target sequences. The speed of calculation depends on the numbers of target sequences and primer pairs involved.
An alternative way to design compatible multiplex PCR primer pairs is to use pre-designed primers as references for the design of new primers. The user can also select input options for the PCR products such as the minimum product size differences between the amplicons. One can set primer design conditions either individually for each given sequence or use common values. The individual setting has a higher priority for PCR primer or probe design than do the general settings. The results include primers for individual sequences, primers compatible together, the product sizes, and annealing temperatures. Because clear differentiation of the products is dependent on using compatible primer pairs in the single reactions, the programme recovers all potential variants of primer combinations for analyses of the chosen DNA regions and provides, in tabular form, their compatibility with information including primer-dimers, cross-hybridisation, product size overlaps, and similar alternative primer pairs based on Tm. The user may choose those alternative compatible primer pair combinations that provide the desired product sizes. Using the programme, researchers can select pre-designed primer pairs from a target for their desired types of PCR reactions by changing the filtering conditions as mentioned above. For example, a conventional multiplex PCR requires differently sized (at least by 10 bp) amplicons for a set of target genes, so the value for the minimum size difference between PCR products can be selected.
In addition to the need to avoid same-sized amplicons, multiplex PCR must also minimise the generation of primer-dimers and secondary products, which becomes more difficult with increasing numbers of primers in a reaction. To avoid the problem of non-specific amplification, jPCR allows the selection of primer pairs that give the most likelihood of producing only the amplicons of the target sequences by choosing sequences which avoid repeats or other motifs. The programme also allows the user to design not only compatible pairs of primers, but also compatible single primers for different targets or sequences.
Group-specific amplification, also called family-specific and sequence-specific amplification, is an important tool for comparative studies of related genes, sequences, and genomes that can be applied to studies of evolution, especially for gene families and for cloning new related sequences. Specific targets such as disease resistance analogues (NBS-profiling) or transposable elements can be amplified to uncover DNA polymorphisms associated with these sequences. The overall strategy of designing group-specific PCR primers uses a hash index of 9-mers to identify common regions in target sequences, following standard PCR design for the current sequence, and then testing complementarity of these primers to the other sequences. In comparison to the software Primaclade [7], Primique [8], UniPrime [6] and GeneFisher [11], jPCR does not use sequence alignment, giving it the flexibility to use a different strategy for primer design. jPCR does not design degenerate PCR primers to amplify a conserved or polymorphic region of all related sequences.
The jPCR package designs large sets of universal primer pairs for each given sequence, identifies conserved regions, and generates suitable primers for all given targets. The steps of the algorithm are performed automatically and the user can influence the general options for primer design options. jPCR will work with any source of sequence as long as it is possible to find short (minimum 12 nT) consensus sequences amongst the sets. The quality of primer design is dependent on sequence relationships, phylogenetic similarity, and suitability of the consensus sequence for the design of good primers. The software is able to generate group-specific primers for each set of sequences independently, which are suitable for all sequences. Primer alignment parameters for group-specific PCR primers are similar to those used for in silico PCR. The software has been experimentally tested extensively for group-specific PCR.
Sequence-independent cloning, including ligation-independent cloning (LIC) [41] requires generation of complementary single-stranded overhangs in both the vector and insertion fragments. Similarly, multiple fragments can be joined or concatenated in an ordered manner using overlapping primers in PCR [42]. Annealing of the complementary regions between different targets in the primer overlaps allows the polymerase to synthesise a contiguous fragment containing the target sequences during thermal cycling, a process called ‘overlap extension PCR’ (OE-PCR) [43]. The efficiency depends on the Tm and on the length and uniqueness of the overlap. To achieve this, the programme designs compatible forward and reverse primers at the ends of each fragment, and then extends the 5′ end of primers using sequences from the primers of the fragment that will be adjacent in the final product. The programme selects the overlapping area so that the primers from overlapping fragments are similar in size and in their optimal annealing temperature. The programme adds the required bases so that the Tm of the overlap is similar to or higher than the Tm of the initial primers. Primers are tested for dimers within the appropriate primer pair.
Several programmes have been developed to automatically design oligonucleotides for long sequence synthesis, based on specification of the oligonucleotide length and Tm threshold [44]; [45] ; [46]. Only the programme TmPrimer [44] provides the prediction of potential interactions between oligonucleotides; however this programme does not consider non-specific hybridisation. Our algorithm is able to design oligonucleotides for long sequences containing repeats and to minimise their potential non-specific hybridisation during 3′ end extension in PCR.
Modelling the hybridisation of primers to targeted annealing sites is the only way to predict PCR products [47]; [48]; [49]; [50]; [51] ; [52]. The last 10–12 bases at the 3′ end of primers are important for binding stability; single mismatches can reduce PCR efficiency, the effect increasing with proximity to the 3′ end. jPCR allows simultaneous testing of single primers or a set of primers designed for multiplex target sequences. It performs a fast, gapless alignment to test the complementarity of the primers to the target sequences. The parameters can be set to allow different degrees of mismatches at the 3′ end of the primers. The programme can also handle degenerate primers or probes including those with 5′ or 3′ tail sequences. Probable PCR products can be found for linear and circular templates using standard or inverse PCR as well as for multiplex PCR. This in silico tool is useful for quickly analysing primers or probes against target sequences, for determining primer location, orientation, efficiency of binding, and calculating their Tm's.
Simple sequence repeats (SSRs, or microsatellites) are short tandem repeats of one or more bases. Microsatellites are ubiquitously distributed throughout eukaryotic genomes, often highly polymorphic in length, and thereby an important class of markers for population genetic studies. Our approach to SSR searching is to analyse low complexity regions by using linguistic sequence complexity. This method allows the detection of perfect and imperfect SSRs with a single, up to 10-base, repeat motif. Each entry sequence is processed for identification of SSRs and the SSR flanks are used to design compatible forward and reverse primers for their amplification by PCR.
The jPCR identifies all SSRs within each entry sequence and designs compatible PCR primer pairs for each SSR locus. The default PCR primer design parameters are that the primers must be within 100 bases from either side of the identified SSR. Often the sequence available around SSR loci is not sufficient for designing good primers and the user can increase or decrease the distance from either side to find more efficient and compatible primer pairs. The jPCR programme has been successfully implemented for designing PCR primers for SSR loci in many laboratories worldwide; some resulting articles are shown on our FastPCR web page [http://primerdigital.com/fastpcr/citations.html]. The capabilities of jPCR make it a complete bioinformatics tool for the use of microsatellites as markers, from discovery through to primer design.
Primer3 and its derived applications, e.g., NCBI/Primer-BLAST [http://www.ncbi.nlm.nih.gov/tools/primer-blast/], IDT SciTools [http://eu.idtdna.com/scitools/, 5], BiSearch web server [http://bisearch.enzim.hu/, 10], Primo Pro 3.4 [http://www.changbioscience.com/primo/] and PerlPrimer [http://perlprimer.sourceforge.net/, 4] are amongst the most commonly used web-based applications. Table 3 and our web page [http://primerdigital.com/tools/soft.html] provide a comparison of features of our primer design and oligonucleotide analysis software to these programmes. We studied, for example, the ability to perform analyses for non-specific amplification in a given sequence. Results for this test for the NCBI/Primer-BLAST and BiSearch web server are available as supplementary materials (S2); generally, other tools do not take repeat regions inside the sequence into account, and can design the primers or probes within them, which may result in non-specific amplification (S2).
Table 3.
Comparison of primer design and oligonucleotide analysis tools.
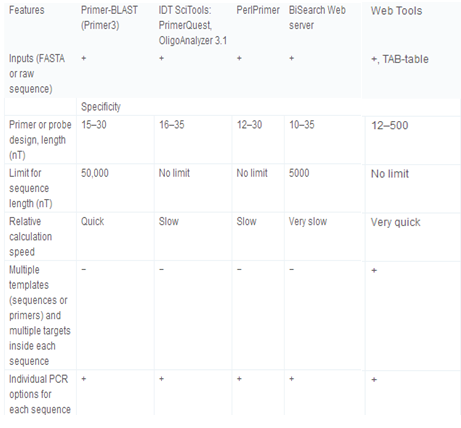
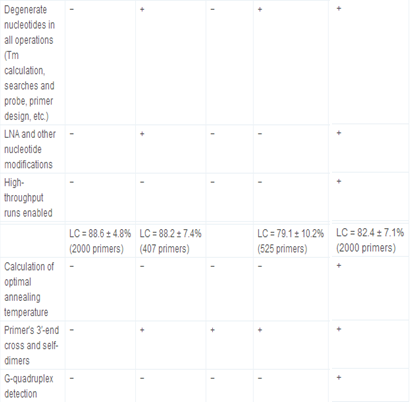
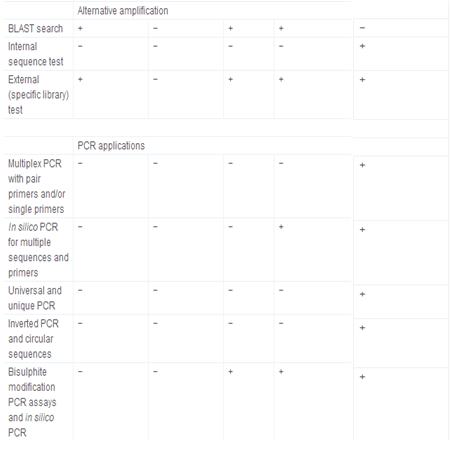
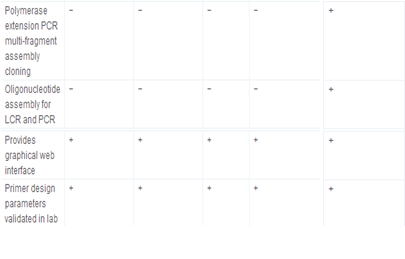
+ feature supported, and −feature not supported.
Primers designed by NCBI/Primer-BLAST for sequences of high CG content (containing 65–70% CG) have 90–100% CG in the terminal 12 bases at the 3′-end. The BiSearch web server and IDT PrimerQuest correctly design primers with a 50% CG content at default conditions. We found that primer amplification efficiency depends not only on the Tm of the primer and its 3′ end sequence, but also its linguistic complexity. For a good primer, the linguistic complexity value must exceed 80%, or 90% for the best primers. The average value for the linguistic complexity of primers for BiSearch web server is 79.1 ± 10.2% (525 primers tested); for NCBI/Primer-BLAST, 82.4 ± 7.1% (2000 primers); for IDT PrimerQuest, 88.2 ± 7.4% (407 primers); for our jPCR, 88.6 ± 4.8% (2000 primers). For example, the primers 5′ TTTTTTTTTGGGGGGGGGAG 3′ (with LC = 38%), polypurine 5′ GGAGAGAGAGAGAAGAGAGAAAG 3′ (LC = 33%), and polypyrimidine 5′ CTCTCTCTCCTCTTTTTCTCC 3′ (LC = 40%), designed with NCBI/Primer-BLAST and BiSearch web servers, risk the formation of stable four-stranded structures.
To compare the efficiency of dimer detection, we designed 2000 primers for chloroplast DNA from Arabidopsis thaliana (1–50,000 bases; AP000423). Using NCBI/Primer-BLAST, we found that 105 primers form unacceptable self-dimers, as shown in some examples ( S3). The BiSearch web server was the slowest web-based software (10 min for 100 primer pairs in 5 kb under default conditions), but the primers it designed were 3′ end dimer-free, only one containing a weak internal self-dimer. Several internal self-dimers (21) and two weak 3′ end dimers were found for primers designed with IDT PrimerQuest (S3). Primers from IDT PrimerQuest showed good linguistic complexity and primer quality values.
We have developed an approach for efficient design of PCR primers and probes, oligonucleotides for long sequence assembly, in silico analyses, and for predicting and analysing oligonucleotide properties. The software can work simultaneously with multiple nucleic acid sequences and internal sequence targets. Our own findings and data from other laboratories have been collected and analysed to predict more precisely oligonucleotide quality for PCR efficiency. Using these to set the parameters for oligonucleotide design, we were able to improve our success rate for PCR. Compared with the tools here, almost all available programmes for designing PCR primers do not investigate the sequence of primer, and use only the primers' melting temperature for selection. The software algorithms can select highly specific oligonucleotides suitable for most PCR applications, with a wide functional temperature range, and can eliminate poor oligonucleotides by using secondary structure prediction.
There are several publicly available tools for designing PCR primers but jPCR is the only one with an integrated tools environment that provides comprehensive facilities for designing primers for most PCR applications, aids in the design of primers simultaneously with multiple nucleic acid sequences as well as internal sequence targets for different PCR applications and their combinations, and allows application of its componentised functionality for a wide range of projects. This software is flexible and allows the application of its componentised functionality for a wide range of projects. It has been tested in numerous laboratories and companies worldwide to design many thousands of PCR primers and probes for a range of applications and tasks [see the citations at http://primerdigital.com/fastpcr/citations.html], providing valuable feedback that has been used to develop the programmes further. We will continue to evaluate the tools through the web of laboratories and collaborators providing feedback.
The tools [http://primerdigital.com/tools/] are written in Java with NetBeans IDE (Oracle) and require the Java Runtime Environment (JRE) on a computer. It can be used with any operating system (64-bit OS preferred for large chromosome files). The Java applications take either a single sequence or accept multiple separate DNA sequences in FASTA or tabulated format (Excel sheet or Word table) or as an RTF file. The PCR primer design algorithm generates sets of primers with a high likelihood of success for use in any amplification protocol. All primers can be used for PCR or sequencing experiments.
The multiplex PCR algorithm is based on the fast non-recursion method, with the software performing checks on product size compatibility and cross-dimer interaction for all primers. For long sequence assembly, oligonucleotide design starts from the 5′ end of a given sequence; the oligonucleotide length is dynamically changed until a unique 3′ end has been found and Tm of oligonucleotide has reached the Tm threshold. All oligonucleotides are designed without gaps between them. The other strand is used for design of the overlapping oligonucleotides with the same algorithm as above but with the Tm of the overlapping regions reaching the Tm — 15 °C threshold. The composition of the sequence at the 3′ terminus is important because stability at the 3′ terminus in the oligonucleotide complexes will improve the specificity of extension by the polymerase. To reduce non-specific polymerase extension and ligation (LCR), the algorithm chooses only unique sequences for the 3′ terminus. Minimally, the last 2 nucleotides at the 3′ terminus must not be complementary to the non-specific target. Other complementary regions, apart from the 3′ terminus, are not important for assembling multiple fragments by PCR and ligation. For in silico PCR, a quick alignment for detection of primer locations on the reference sequence is performed by analyses on both strands using a hash index of 3- or 7-mers (containing up to one mismatch within) and by calculating the local similarity for the whole primer sequence. The parameters for quick alignment may be set: the minimum is 3 bases for search initiation and 50% local similarity.
The programme generates primer pairs (and probes) from the input sequences and shows the optimal annealing temperature for each primer pair and the sizes of PCR products together with information for each designed primer. Results are generated by the programme showing the suggested primers and primer pairs in tabulated format for Excel or Open Office. The spreadsheets show the following properties: automatically generated primer name, primer sequence, sequence location, direction, length, melting temperature, CG content (%), molecular weight, molar extinction coefficient, linguistic complexity (%) and PQ. For compatible primer pairs, the annealing temperature and PCR product size are also provided.
Linguistic complexity values are converted to percentages, 100% being the highest level:
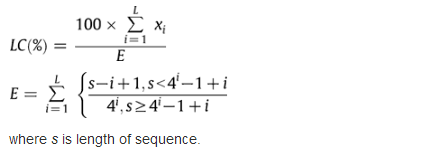
For example, the sequence 5′-ACACACACACACACAC, 16 nT, contains two nucleotides (A, C), but expected E = 4 variants; two variants of two-nucleotides (AC, CA), but expected E = (16 − 1) variants; two variants of three-nucleotides (ACA, CAC), and expected E = (16 − 2) variants. The complexity value is LC = 100 ∗ (2 + 2 + 2) / (4 + 16 − 1 + 16 − 2) = 18.2%.
Intermolecular G-quadruplex-forming sequences are detected according to the formula …Gm1XnGm2…, where m is the number of G residues in each G-tract (m1, m2 ≥ 3); the gap Xn (n ≤ 2 ∗ minimal (m1:m2)) can be any combination of residues, including G [25]. The gap sequences (Xn) may have varying lengths, and a relatively stable quadruplex structure may still be formed with a loop more than 7 bases long, but in general, increasing the length of the gap leads to a decrease in structure stability. It is also possible for one of the gaps to be zero length when there are long poly-G tracts of > 6 bases.