

发布者:Primerbank 时间:2017-09-25 浏览量:1540
Bioinformatics, Volume 31, Issue 9, 1 May 2015, Pages 1472–1474, https://doi.org/10.1093/bioinformatics/btu832
Published: 17 December 2014 Article history

Primer design is complicated by many factors, including desired primer specificity or promiscuity, restrictions in polymerase chain reaction (PCR) or sequencing lengths, matching of melting temperatures, heterogeneous evolutionary rates across genomes, avoiding primer–primer dimerization and integrating bio-barcodes. Typically, the process of finding the optimal placements for primers is a serial process in which each of the design criteria are optimized one at a time. This is unfortunate as one design step may influence another. Thus, designing primers while optimizing all design criteria simultaneously is desirable. Because no existing software was able to do that, we recently developed a tool that could include all these factors, given a multiple-alignment and user input on desired constraints, to design primer pairs for single-fragment analyses (Brodin et al., 2013). However, with the increased abilities of next-generation sequencing (NGS) a need to design optimal multiple-fragment analyses, such as tiled fragments that cover entire small genomes, has rapidly emerged.
Many previous primer design software have focused on special purposes such as multiplexing, degenerate sites, discriminate amplification, nested PCR, single-nucleotide polymorphism (SNP) protocols, and hybridization analyses such as micro-arrays and in situ hybridization, and DNA sequencing (Fredslund et al., 2006; Giegerich et al., 1996; Kalendar et al., 2011; Rouillard et al., 2003; Rychlik 2007; Untergasser et al., 2012; Vallone and Butler, 2004; Weckx et al., 2005). More generic software, such as Primer3 (Untergasser et al., 2012), base their design on a single sequence, which inadvertently could place primers in more variable regions, leading to annealing failure with some templates or selective amplification in diverse populations.
Motivated by the large genetic diversity found in Human immunodeficiency virus (HIV) and Simian immunodeficiency virus (SIV), and the need for a generic design tool that could dynamically optimize primers across their genomes, we developed PrimerDesign-M. It can be used to design single- or multiple-fragment primers to any aligned set of DNA sequences, regardless of diversity level and organism.
The input consists of a multiple-alignment and user-specified primer design criteria, uploaded and entered into a web interface. The alignment can either be user provided or directly imported from the premade HIV or SIV gene or genome alignments available at the Los Alamos HIV sequence database. To avoid placing primers in gappy regions, gaps may be removed at a user-provided cut-off.
The user selects a region of interest (ROI), which defines the part of the alignment that must be covered by the primers. Note that the alignment must be longer than the ROI so that flanking primers can be designed. Next, the user chooses whether a single pair or multiple pairs of primers should cover the ROI. Adaptors can be added, and expected experimental read lengths are defined. If multiple-primer design is selected, five different design options become available: Max for maximum overlap between fragments, useful when one wants to ensure contig coverage even when some fragments may fail, Mid for a ‘tiled’ fragment design, Min for minimum overlap, Zero for no overlap, i.e. 3′-primers are reused as 5′-primers in next fragment and Flex for moving across the ROI placing forward primers in positions with as low as possible entropy scores anywhere within the previous fragment.
DNA barcode tags can be added, optimizing either number of tags, tag length or tag edit distance. Additional design options include primer lengths, primer detection limit (design inclusion of degenerated primer sites to cover genetic diversity), complexity limit (maximum number of primer species caused by degenerated sites), maximum difference in Tm between primers, G/C clamps and dimerization risk [dimer window size to be considered, and maximum ratio of matched nucleotides within the dimer window; the default dimerization values are based on experimental results (Desmarais et al., 2012)]. The dimerization test includes all possible homo- and hetero-dimer formations, including hairpin structures. Tm’s are calculated using the empirical nearest neighbour model (SantaLucia et al., 1996). For designs that may take more than a minute to design, the user inputs a mail-back email address.
All input options have a link that explains parameter definitions and value ranges. As usual in HIV database tools, we also provide example input to test run the tool.
The output consists of a summary of design criteria, one or several design graphic representations (depending on if single- or multiple-fragment design was chosen), tables with primer designs, and download options for graphics, tables, gap-stripped alignment and dimerization test logs. We provide five alternative primer pairs per fragment to allow troubleshooting and potential alternatives, should one set experimentally fail. The list of primer pairs is ordered by least entropy among primers that fulfill all design criteria. This should make it easy to continue with an otherwise successful design. Graphics are available in png, pdf and jpg formats, and tables can be downloaded in txt format to be read by spreadsheet or database programs. For multiple- fragment design, a summary graphic shows the overall design including the ROI and the locations of the first alternative set of primers per fragment, curves for entropy, complexity and Tm (Fig. 1).
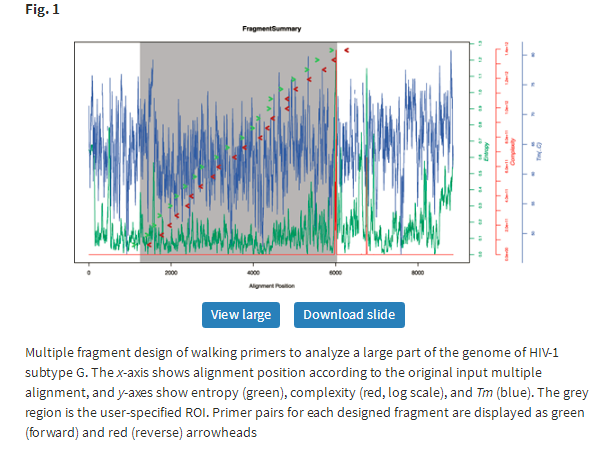
If mail-back was used, the link to the results stays active for 4 days. If the design fails at some point during multiple-fragment optimization, the output will still show successful fragment design up until the failed fragment. This should allow the user to modify the design criteria that can cover the whole ROI or define a new ROI for the remaining sequence.
Figure 1 shows a potential walking-primer design to amplify/sequence HIV-1 subtype G. Here, we used the HIV database ‘web alignment’ of 2013, and a ROI in positions 2000–7000 with gaps removed if more frequent than 50% in any alignment column. We used the Flex fragment overlap option as we wanted to make sure primers sit in regions with as low diversity as possible given other constraints, especially read length; we wanted to use custom sequencing adaptors (‘My adaptor’ input was M13 forward adaptor CGCCAGGGTTTTCCCAGTCACGAC and reverse adaptor AGCGGATAACAATTTCACACAGGA); an expected read length 300–400 nt; no barcode tags; and default settings otherwise (minimum primer length = 20, maximum primer length = 25, detection limit = 5%, complexity limit = 32, maximum difference in Tm = 5°C, dimer window size = 10, dimer max ratio = 0.9, G/C clamp on forward and reverse primers).
The design resulted in 21 fragments 300–400 nt long covering the ROI, which was modified to positions 1242–6032 when gaps were removed. The resulting gapstripped alignment could be downloaded. For each of the 21 fragments a table with five alternative pairs was provided. It took about 2 min for PrimerDesign-M to complete this design.
PrimerDesign-M can design primers for both single fragment analyses as well as multiple-fragment walking-primer analyses of highly variable genomes, and the designed primers have been experimentally confirmed to work (Brodin et al., 2013; Fischer et al., 2010). PrimerDesign-M should be useful to researchers in all fields of biology who need to design DNA primers. To make access easy, guarantee that the latest version of the tool is used and make the program independent of computer platform, the software is available as a web tool at the LANL HIV database.
This project was funded by a National Institutes of Health - Department of Energy (NIH-DOE) interagency agreement [Y1-AI-8309] and National Institutes of Health (NIH) [grant R01AI087520].