

发布者:Primerbank 时间:2017-09-29 浏览量:3641
一.实验目的:
SDS-PAGE不仅可以分离鉴定蛋白质,而且可以根据迁移率大小测定蛋白质亚基的分子量。
二.SDS-PAGE原理
SDS电泳技术首先在1967年由Shapiro建立,1969年由Weber和Osborn进一步完善。当在样品介质和聚丙烯酰胺凝胶系统中加入SDS后,则蛋白质分子的电泳迁移率主要取决于它的分子量大小,其它因素可以忽略不计。
SDS是一种阴离子去污剂,它能破坏蛋白质分子之间以及其它物质分子之间的非共价键。在强还原剂如巯基乙醇或二硫苏糖醇的存在下,蛋白质分子内的二硫键被打开并解聚成多肽链。解聚后的蛋白质分子与SDS充分结合形成带负电荷的蛋白质-SDS复合物,复合物所带的负电荷大大超过了蛋白质分子原有的电荷量,这就消除了不同蛋白质分子之间原有的电荷差异,蛋白质-SDS复合物在溶液中的形状像一个长椭圆棒。椭圆棒的短轴对不同的蛋白质亚基-SDS复合物基本上是相同的,但长轴的长度则与蛋白质分子量的大小成正比,因此这种复合物在SDS-PAGE系统中的电泳迁移率不再受蛋白质原有电荷的影响,而主要取决于椭圆棒的长轴长度即蛋白质及其亚基分子量的大小。当蛋白质的分子量在15-200kD之间时,电泳迁移率与分子量的对数呈线性关系。
SDS-PAGE凝胶的有效分离范围
丙烯酰胺浓度(%) 15 12.5 10 7.5 5.0
线性分离范围 15-43 kDa 15-60 kDa 18-75 kDa 30-94 kDa 60-212 kDa
三.实验步骤:
1)溶液配置:
1.分离胶缓冲液1.5M Tris-HCL (pH8.8)100ml:
18.17g Tris-base,50ml蒸馏水,加入浓盐酸调pH8.8,最后定容至100ml。4℃贮存备用。
2.积层胶缓冲液1M Tris-HCL (pH6.8)100ml:
12.1g Tris-base,50ml蒸馏水,加入浓盐酸调pH6.8,最后定容至100ml。4℃贮存备用。
3.10%SDS,100ml:
10gSDS,加入50ml蒸馏水在60℃搅拌溶解,待泡沫消失后,定容至100ml。
4.30%聚丙酰胺贮液(A液)100ml:
丙烯酰胺29g,甲叉丙烯酰胺1g,去离子水溶解,定容到100ml。0.45μm滤器过滤除菌,4℃棕色瓶保存。pH≤7.0。
5.考马斯亮蓝染色液 100ml:
考马斯亮蓝R-250 0.25g 甲醇45ml ,冰醋酸 10ml, 蒸馏水45ml
6.考马斯亮蓝脱色液 100ml:
甲醇45ml,冰醋酸10ml,蒸馏水45ml
7.SDS电泳缓冲液(25nM Tris 碱,250mM甘氨酸,PH8.3 0.1%SDS)
试剂 MW 所需量 终浓度
Tris 碱 121.1 15.1g 125mM
甘氨酸 75 94g 1.25M
10%SDS 50ml 0.5%
注意,在加入SDS前应测定PH值
8.10%过硫酸铵:0.1g过硫酸铵溶解于1ml纯水中。
11.2×SDS凝胶上样缓冲液:
试剂 所需量 终浓度
1Mtris-HCl(PH6.8) 1.0ml 100mM
Beta-巯基乙醇(或者1M二硫苏糖纯) 1.0ml(2.0ml) 10%(200mM)
10%SDS 4.0 ml 4%
甘油 2.0ml 20%
溴酚兰 20mg 0.2%
分装后,-20℃保存
2)实验操作:
1. 样品处理:将样品加入等量的2×SDS上样缓冲液,100℃加热3-5分钟,离心12000×2分钟,取上清做SDS-PAGE分析,同时将分子量蛋白标准样品做平行处理。
2. 按说明书转好垂直式电泳槽装置
要注意:安装前,胶条、玻板、槽子都要洁净干燥;勿用手接触灌胶面的玻璃。
3.配胶:根据所测蛋白质分子量范围,选择适宜的分离胶浓度。按下表的配制分离胶和浓缩胶。
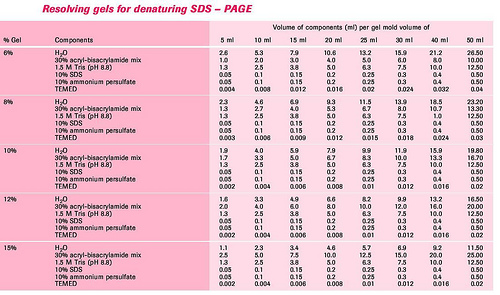
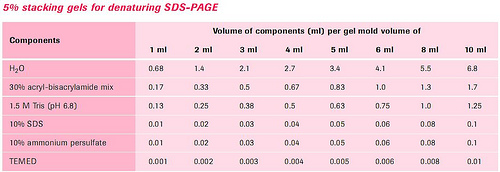
制备凝胶板:
(1)分离胶制备:按表配制分离胶,混匀后用细长头滴管将凝胶液加至长、短玻璃板间的缝隙内,约8cm高,用1ml注射器取少许蒸馏水,沿长玻璃板板壁缓慢注入,约3—4mm高,以进行水封。约30min后,凝胶与水封层间出现折射率不同的界线,则表示凝胶完全聚合。倾去水封层的蒸馏水,再用滤纸条吸去多余水分。
(2)浓缩胶的制备:按表配制浓缩胶,混匀后用细长头滴管将浓缩胶加到已聚合的分离胶上方,直至距离短玻璃板上缘约0.5cm处,轻轻将样品槽模板插入浓缩胶内,避免带入气泡。完全聚合需15-30min.待凝胶凝固,小心拔去样品槽模板,用窄条滤纸吸去样品凹槽中多余的水分,将1×电泳缓冲液倒入上、下贮槽中,应没过短板约0.5cm以上,即可准备加样。
4.加样
一般加样体积为10-30μL(即2-10μg蛋白质)。如样品较稀,可增加加样体积。用微量注射器小心将样品通过缓冲液加到凝胶凹形样品槽底部,待所有凹形样品槽内都加了样品,即可开始电泳。
5.电泳
将直流稳压电泳仪开关打开,积层胶电压8×V/cm,分离胶电压15v/cm,先80v30分钟,待样品进入分离较时,在150v2小时。当蓝色染料迁移至底部时,将电流调回到零,关闭电源。拔掉固定板,取出玻璃板,用刀片轻轻将一块玻璃撬开移去,在胶板一端切除一角作为标记,将胶板移至大培养皿中染色。
6.染色及脱色
将染色液倒入培养皿中,室温染色4h以上,用蒸馏水漂洗数次,再用脱色液脱色,多次脱色直到蛋白带清晰。
7.凝胶摄像和保存
在图像处理系统下将脱色好的凝胶摄像,结果存于软盘中,凝胶可保存于双蒸水中或70%乙酸溶液中。
四.注意事项
1. 不是所有的蛋白质都能用SDS-凝胶电泳法测定其分子量,已发现有些蛋白质用这种方法测出的分子量是不可靠的。包括:电荷异常或构象异常的蛋白质,带有较大辅基的蛋白质(如某些糖蛋白)以及一些结构蛋白如胶原蛋白等。例如组蛋白F1,它本身带有大量正电荷,因此,尽管结合了正常比例的SDS,仍不能完全掩盖其原有正电荷的影响,它的分子量是21,000,但SDS-凝胶电泳测定的结果却是35,000。因此,最好至少用两种方法来测定未知样品的分子量,互相验证。
2.有许多蛋白质,是由亚基(如血红蛋白)或两条以上肽链(如α-胰凝乳蛋白酶)组成的,它们在SDS和巯基乙醇的作用下,解离成亚基或单条肽链。因此,对于这一类蛋白质,SDS-凝胶电泳测定的只是它们的亚基或单条肽链的分子量,而不是完整分子的分子量。为了得到更全面的资料,还必须用其它方法测定其分子量及分子中肽链的数目等,与SDS-凝胶电泳的结果互相参照。
3.实验组与对照组所加总蛋白含量要相等
4.为达到较好的凝胶聚合效果,缓冲液的ph值要准确,10%过硫酸铵在一周内使用。室温较低时,TEMED的量可加倍。
5.未聚合的丙烯酰胺和亚甲双丙烯酰胺具有神经毒性,可通过皮肤和呼吸道吸收,应注意防护。
五.SDS-PAGE trouble-shooting
1) 开始电泳时没有电流:
可能原因:上槽或下槽的电泳缓冲液体积不够。
解决的方法:确信上下槽的电极缓冲液中有足够的缓冲液,同时检查是否漏液。
2)在分离胶中样品进行缓慢:
可能原因:SDS电泳缓冲液配置错误,或是分离胶缓冲液配置错误,或是丙烯酰胺溶液不新鲜造成。
解决方法:重新配置新鲜的缓冲液
3)染料的底部不平(微笑现象)
可能的原因:胶没有冷却均匀或是电流太高
解决方法:1.在电泳过程中,使用循环水冷却胶,同时在下槽缓冲液加一搅拌子,使液体温度均匀。2.下槽缓冲液的体积尽可能大3.限制电流的大小
4)皱眉现象
可能原因:在上样梳的位置胶没有聚合好,装置的安装不合适,或是上槽缓冲液漏液。
解决的方法:1.给凝胶溶液脱气或将过硫酸铵和TEMED的浓度加大一半。2.确定仪器的垫圈没有被压住3.确信有足够量的上槽缓冲液。
5).染料带形不规则:
可能原因:凝胶的均一性很差,或胶的顶部不平造成。
解决的方法:1.给凝胶溶液脱气或将过硫酸铵和TEMED的浓度加大一半2.在倒完胶后,立刻在胶的表面覆盖上一层溶液把胶的表面压平。