

发布者:Primerbank 时间:2017-09-29 浏览量:3608
回顾分子生物学的发展历程,PCR技术的发明和普及无疑是最重要的篇章之一。而PCR技术在近20年的不断发展创新中,最受瞩目当属荧光实时定量PCR技术(real-time quantitative PCR, or qPCR)。定量PCR技术真正实现了PCR从定性到定量的飞跃,通过对PCR过程的实时监控,专一、灵敏快速、可重复地精确定量起始模版浓度,已经在科研和临床诊断领域得到了越来越广泛的应用。本文从荧光定量PCR的原理入手,详细介绍荧光定量PCR仪的类型和技术,为定量PCR仪的选择提供详细参考。
一、背景
本来,PCR是为了将样本中微量的DNA模版放大以便研究模版特性,随着研究的深入,要了解样本中基因的表达模式与疾病的关系,就需要了解标本中的DNA原始拷贝数。理论上PCR过程中反应产物是以指数规模增长的,但实际的PCR扩增曲线并不是标准的指数曲线,而是S形曲线——因为随着PCR循环数的增加,扩增规模迅速增大,Taq酶、dNTP、引物,甚至DNA模板等各种PCR要素逐渐不敷需求,PCR的效率降低,产物生成的速度逐渐减缓,最终进入平台期。由于各种环境因素的复杂相互作用,不同的PCR反应体系进入平台期的时间和平台期的高低都有很大变化,即使是重复实验,各种条件基本一致,最后得到的DNA拷贝数也往往不同。因此经过PCR扩增的DNA产物量不能反映起始模板量的真实情况。通过传统凝胶电泳EB染色或者同位素标记只能定量PCR的终产物量,而不能定量起始DNA模版的拷贝数。
荧光实时定量PCR技术是指在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。借助于荧光信号来检测PCR产物,一方面提高了灵敏度,另一方面可以在每次PCR循环收集数据,建立实时扩增曲线,准确地确定域值循环数(CT值),计算起始DNA拷贝数,做到了真正意义上的DNA定量。根据最终得到的数据不同,定量PCR可以分为相对定量和绝对定量两种。相对定量常见于比较两个样本中基因表达水平的高低变化,得到的结果是百分比;绝对定量则需要使用标准曲线确定样本中基因的拷贝数或浓度。
荧光实时定量PCR技术最早是在1992年由一位日本人Higuchi第一次报告提出的,他当时的初衷很简单,就是想实时地看到PCR反应的整个过程,由于当时EB(溴化乙锭)的广泛使用,最直接想到的标记染料就是EB,可以插入到双链核酸中受激发光。这样,在普通PCR仪的基础上再配备一个激发和检测的装置,第一台可实时监测的PCR仪就诞生了。在PCR反应的退火或延伸时检测掺入到双链核酸中EB的含量就能实时监控PCR反应的进程,考虑到PCR反应的数学函数关系,结合相应的算法,通过加入标准品的方法,就可以定量待测样品中的目标基因。定量PCR技术的提出不单是PCR技术的飞跃,也DNA定量技术的一次飞跃。经过不断发展完善,到1996年当时的PE公司(后来的ABI)推出了世界第一台商品化的荧光定量PCR仪,标记方法也由最初的染料法,逐渐发展到了特异性更高的Taqman探针法,以及Molecular Beacon、Fret等不同方法的标记探针。由于ABI公司在PCR领域的领袖地位和推动定量PCR技术的早期发展的特殊贡献, Taqman探针法得到广泛使用。
二、常用荧光标记方法
常用的荧光标记方法可简单分为两大类:1.非特异检测——双链DNA内插式荧光染料;2.扩增序列专一检测——主要指荧光探针和引物探针。荧光染料技术成本低廉,实验设计简便;而探针杂交技术在原理上严格,所得数据特异性高、更为精确。在选择实验方案时要根据实验目的和对数据精度的要求来决定。
非特异性检测扩增序列的代表是SYBR Green I荧光染料。SYBR Green I只与DNA双链结合,插入DNA双链时发出荧光;DNA双链解链时释放出来,荧光信号急剧减弱。在一个加入过量SYBR green 1荧光染料的体系内,其信号强度代表了双链DNA分子的数量。SYBR Green荧光染料法定量PCR的基本过程是:1、开始反应,当SYBR Green染料与DNA双链结合时发出荧光。2、DNA变性时,SYBR Green染料释放出来,荧光急剧减少。3、在聚合延伸完成后,SYBR Green染料与双链产物结合,定量PCR系统检测到荧光的净增量加大。SYBR Green I 的最大吸收波长约为497nm,发射波长最大约为520nm。
荧光染料的优势在于能监测各种双链DNA序列的扩增,无需设计探针,检测简单简便,成本低廉。然而正是由于荧光染料能和任何dsDNA结合,对DNA模板没有选择性,因此它也能与非特异的dsDNA(如引物二聚体)结合,使实验容易产生假阳性信号,引物二聚体的问题目前可以用带有熔解曲线(melting curve)分析的软件加以解决。要想用荧光染料法得到比较好的定量结果,对PCR引物设计的特异性和PCR反应的质量要求就比较高。
荧光探针法是用序列特异的荧光标记探针来检测产物,探针法的出现使得定量PCR技术的特异性比常规PCR技术大大提高。目前较常提及的有TaqMan探针、FRET杂交探针(荧光共振能量传递探针)和分子信标Molecular Beacon。
广泛使用的TaqMan探针法是指PCR扩增时在加入一对引物的同时另外加入一个特异性的荧光探针,该探针只与模板特异性地结合,其结合位点在两条引物之间。探针的5′端标记有荧光报告基团(Reporter, R),如FAM、VIC等,3′端标记有荧光淬灭基团(Quencher, Q),如TAMRA等。当探针完整的时候,5′端报告基团经仪器光源激发的荧光正好被近距离的3′端荧光基团淬灭,仪器检测不到5′端报告基团所激发的荧光信号(就是说5’荧光基团的发射波长正好是3’ 荧光基团的吸收波长,因而能量被吸收传递到3’荧光基团而发出其它荧光)。随着PCR的进行,Taq酶在链延伸过程中遇到与模板结合的探针,其5′-3′外切酶活性(此活性是双链特异性的,游离的单链探针不受影响)就会将切割探针,释放5′端报告基团游离于反应体系中,远离3′端荧光淬灭基团的屏蔽,5′端报告基团受激发所发射的荧光信号就可以被探头检测到。也就是说每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。报告信号的强度就代表了模板DNA的拷贝数。
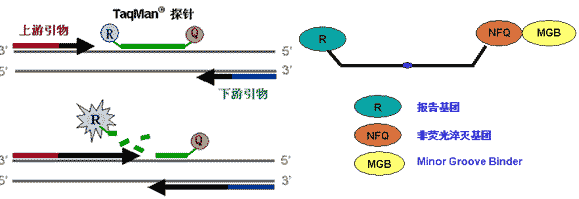
Taqman探针检测的是积累荧光。常用的荧光基团有FAM,TET,VIC,HEX等等。当探针完整的时候,由于3′端的荧光淬灭基团在吸收5′端报告基团所发射的荧光能量,本身会发射波长不同的荧光而导致本底高,因此TaqMan探针近来又有新的发展——TaqMan MGB探针。MGB探针的淬灭基团采用非荧光淬灭基团(Non-Fluorescent Quencher),本身不产生荧光,可以大大降低本底信号的强度。同时探针上还连接有MGB (Minor Groove Binder)修饰基团,可以将探针的Tm值提高10°C左右。因此为了获得同样的Tm值,MGB探针可以比普通TaqMan探针设计得更短,既降低了合成成本,也使得探针设计的成功率大为提高——因为在模板的DNA碱基组成不理想的情况下,短的探针比长的更容易设计。实验证明,TaqMan MGB探针对于富含A/T的模板可以区分得更为理想。
Taqman探针法已经得到广泛使用,不过有人认为这种技术利用了Taq酶5`—3`外切酶活性,一般试剂厂家只给Taq酶的聚合酶活性定标,没有同时给Taq酶5`—3`外切酶活性定标,不同批号试剂之间会给定量带来差异。另外对探针的熔点温度(Tm)仅要求其高于60°C,这就使不同试剂盒之间的特异性参差不齐,难于做质控检测。
FRET探针是罗氏的专利,又称双杂交探针,FRET探针由两条相邻探针组成,在上游探针的3`端标记供体荧光基团(例如FAM),下游探针的5`端标记受体荧光基团(如Red 640)。当PCR变性时两探针处于游离状态,供体荧光分子受激发产生的荧光因为距离远而不能被受体荧光基团吸收,受体荧光基团不发光,探头就检测不到指定波长荧光。当PCR退火时,两探针同时结合在模板上,供体基团和受体荧光素相邻,供体荧光素受激发而产生的荧光正好被邻近的受体荧光基团吸收而发出另一波长的荧光,检测探头所检测的正是这个受体基团发射的荧光,这个过程称为荧光能量共振传递(fluorescence resonance energy transfer,FRET)。由于FRET探针是靠近发光,所以检测信号是实时信号,而非累积信号。FRET探针由于是双杂交探针,特异性更高,但是成本也比较高。另外还可利用荧光寡核苷酸熔解曲线(melting curve)对与寡核苷酸探针结合的序列进行分析,从中获取有用的信息。常用的荧光基团是:LC-Red640,LC-Red705。
分子信标(molecular beacon)是一类能形成发夹结构(或者叫茎环结构)的探针,序列两末端序列互补(5—8bp左右),中间环一般为15-30个核苷酸长,并与目标序列互补。探针一端标记报告荧光基团,另一端结合淬灭基团,当探针游离呈发夹结构时,结合在其两端的荧光基团距离上接近,报告基团激发的荧光能量被淬灭基团吸收而使检测探头无法检测到。当互补序列出现时,探针与DNA杂交,探针转变成一个开放的线性结构,报告荧光基团与淬灭荧光基团彼此在空间上产生足够的分离,荧光基团脱离了淬灭基团的影响,从而产生可被检测到的荧光。分子信标探针要求特定结构的序列,设计时必须非常仔细——在复性温度下,模板不存在时形能成茎环结构,模板存在时则与模板配对。并非所有模版都能找到合适的分子信标探针序列。
不过这个概念引申出来就产生了荧光标记引物——把荧光基团标记的发夹结构的序列直接与PCR引物相结合,使引物同时起到荧光探针的作用,从而使荧光标记基团直接掺入PCR扩增产物中。蝎子引物(scorpion primers)和Amplifluor引物都是在这个设计的延伸。蝎子引物由分子信标探针与一段引物序列连接而成。在未杂交状态下,探针由于自身配对形成发夹结构,使荧光淬灭。在PCR反应过程中,随着引物的延伸,探针与新合成链上的靶序列进行分子内杂交,发夹结构打开,产生荧光信号。由此荧光信号的变化即可对原始模板进行定性或定量分析。Amplifluor系统使用一个通用的引物,包含一个18碱基的序列(“Z 序列”),“Z 序列”互补形成发夹结构,并标有荧光基团与淬灭剂。在目标序列的一对特异性引物其中一个5’端加有Z序列,用这对引物扩增目标序列时,通用引物与Z序列配对并进行扩增,发夹结构的打开,使得荧光基团与淬灭剂分离,因此发射的荧光信号的强度与扩增产物的数量成正比。使用这类引物不需要专门设计探针,也不要求模版具有特定的序列,即省了成本又给实验设计提供了宽松的条件。当然由于省略了特异性探针,其专一性不如荧光探针法。
三、定量原理
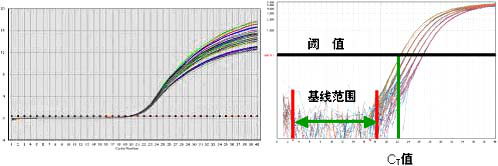
定量PCR可以实时检测PCR的全过程,为了准确定量和计算,取PCR反应的前半段指数扩增期数据。因为检测的是荧光信号,首先需设定一个域值(threshold)作为荧光本底信号(baseline)。从扩增曲线上可以看到,在初始的若干个循环,由于荧光信号变化弱而一直在本底基线附近波动。当检测到的荧光信号超过域值,就被认为是真正的信号,它可用于定义样本的域值循环数(Ct)——PCR扩增过程中荧光信号开始由本底进入指数增长阶段的拐点所对应的循环次数。实验操作中,Ct值的含义是:每个反应管内的荧光信号达到设定的域值时所经历的循环数。
显然,CT值取决于域值。域值的量化定义是基线范围内荧光信号强度标准偏差的10倍。实验操作中,域值范围定义是从第3个循环起到CT值前3个循环止,其终点要根据每次实验的具体数据调整(前2个循环荧光信号太弱,扣除背景后的校正信号往往波动比较大;而在CT值前3个循环大多数情况下荧光信号已经开始增强,超过了基线高度)。从图1的重复实验中可以直观地看到,随着PCR反应过程,荧光信号从基线经一个转折点进入指数期、线性期和最终的平台期,尽管平台期DNA拷贝数波动很大,CT值却是相对固定的。如果用不同浓度模版DNA作PCR,可以看出模版浓度越高,CT值越小。模版浓度每增加1倍,CT值减小1个循环。CT值与模板DNA的起始拷贝数成反比。这一结论可以从数学上严格证明。利用已知起始拷贝数的标准品可作出标准曲线,因此只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
模板定量可分为相对定量和绝对定量。绝对定量指的是用已知量的标准品作标准曲线来推算未知的样本的量。质粒DNA常作为绝对定量标准品的制备之用。标准品的量可根据260nm的吸光度值并用DNA或RNA的分子量来转换成其拷贝数来确定。相对定量指的是在样本中目标序列相对于另一参照样本的量的变化。即参照物是1*的样本,其它的样本为参照物量的n倍。可以通过标准曲线法或者比较CT法进行相对定量,前者和绝对定量相似,后者运用了数学公式来计算相对量,前提是假设每个循环增加一倍的产物数量,在PCR反应的指数期得到CT值来反应起始模板的量,一个循环(CT=1)的不同相当于起始模板数2倍的差异。但是此方法是以靶基因和内参照的扩增效率基本一致为前提的,效率的偏移将影响实际拷贝数的估计。
无论绝对定量还是相对定量,在得到实验结果后,还要考虑数据之间的可比性问题。在实验操作中,取样都是以体积或重量为单位的,但是同样体积或重量的样本所来源的细胞数目并不一样,所以拷贝/μL或拷贝/ng的定量数据相互之间实际上并不可比。只有将这些数据归一到以拷贝/细胞或拷贝/基因组为单位后,才可进行严格意义上的比较。这种校正可以通过适当的参比来完成。参比一般选用b-actin、GAPDH、rRNA基因等管家基因。由于它们在细胞中的表达量或在基因组中的拷贝数是恒定的,受环境因素影响较小,其定量结果代表了样本中所含细胞或基因组的数量。为了减少误差,目标基因和参比基因最好在同一反应管内同时进行定量测定,所以这种对照称为阳性内对照(Internal Positive Control,IPC)。要进行IPC归一化校正,定量PCR仪必须具备多色检测能力,最好是4色。否则,目标基因和参比基因只能分两管作定量,就不成其为“内”标了。
定量实验与定性实验最大的不同,是要考虑统计学要求并对数据进行严格的校正,以消除偶然误差。因此重复实验和设立各种对照非常重要。这一点在实践中往往被轻视或忽视。定量实验,误差是不可避免的。设立重复实验,对数据进行统计处理,可以将误差降低到最小。所以定量实验的每个样本至少要重复3次以上,严格的定量更应当重复6~8次,以满足小样本统计的要求。 如果作绝对定量,则标准曲线需要在5个点以上。标准曲线使用的标准品是浓度已知的DNA样本,虽然可以自己制备,为标准化以及发表文章起见最好购买商品化的标准品。标准曲线的反应条件应当与样本完全一致,以便准确定量。