

发布者:Primerbank 时间:2017-09-30 浏览量:1053
Jason D. Gans John Dunbar Stephanie A. Eichorst La Verne Gallegos-GravesMurray Wolinsky Cheryl R. Kuske
Nucleic Acids Research, Volume 40, Issue 12, 1 July 2012, Pages e96,https://doi.org/10.1093/nar/gks238
Published: 20 March 2012 Article history
Environmental biosurveillance and microbial ecology studies use PCR-based assays to detect and quantify microbial taxa and gene sequences within a complex background of microorganisms. However, the fragmentary nature and growing quantity of DNA-sequence data make group-specific assay design challenging. We solved this problem by developing a software platform that enables PCR-assay design at an unprecedented scale. As a demonstration, we developed quantitative PCR assays for a globally widespread, ecologically important bacterial group in soil, Acidobacteria Group 1. A total of 33 684 Acidobacteria 16S rRNA gene sequences were used for assay design. Following 1 week of computation on a 376-core cluster, 83 assays were obtained. We validated the specificity of the top three assays, collectively predicted to detect 42% of the Acidobacteria Group 1 sequences, by PCR amplification and sequencing of DNA from soil. Based on previous analyses of 16S rRNA gene sequencing, Acidobacteria Group 1 species were expected to decrease in response to elevated atmospheric CO 2 . Quantitative PCR results, using the AcidobacteriaGroup 1-specific PCR assays, confirmed the expected decrease and provided higher statistical confidence than the 16S rRNA gene-sequencing data. These results demonstrate a powerful capacity to address previously intractable assay design challenges.
Environmental biosurveillance and microbial ecology studies use PCR-based assays to detect and quantify microbial taxa and gene sequences within a complex background of microorganisms. However, the fragmentary nature and growing quantity of DNA-sequence data make group-specific assay design challenging. We solved this problem by developing a software platform that enables PCR-assay design at an unprecedented scale. As a demonstration, we developed quantitative PCR assays for a globally widespread, ecologically important bacterial group in soil,Acidobacteria Group 1. A total of 33 684 Acidobacteria 16S rRNA gene sequences were used for assay design. Following 1 week of computation on a 376-core cluster, 83 assays were obtained. We validated the specificity of the top three assays, collectively predicted to detect 42% of the Acidobacteria Group 1 sequences, by PCR amplification and sequencing of DNA from soil. Based on previous analyses of 16S rRNA gene sequencing, Acidobacteria Group 1 species were expected to decrease in response to elevated atmospheric CO 2 . Quantitative PCR results, using the AcidobacteriaGroup 1-specific PCR assays, confirmed the expected decrease and provided higher statistical confidence than the 16S rRNA gene-sequencing data. These results demonstrate a powerful capacity to address previously intractable assay design challenges.
Polymerase chain reaction (PCR)-based assays to detect and quantify microbes in environmental samples containing a complex background of microbial DNA are important tools in national defense, public health and microbial ecology. Consequently, many algorithms have been developed for PCR-assay design. Existing algorithms can be divided into four categories based on the number of target and related non-target sequences that are evaluated during design: (i) a single target sequence, (ii) multiple target sequences , (iii) a single target sequence and multiple non-targets and (iv) multiple target sequences and multiple non-targets . The last category is the most general and most challenging problem—the group-specific assay design problem.
As more sequence data is deposited in public databases, the number of groups that can be monitored increases. Target groups include pathogens and their closest relatives , functional groups or broad taxonomic groups . However, the growing amount of sequence data presents substantial challenges for assay design. Three central challenges are (i) the variation in the length and overlap of available sequences, (ii) the absence of target-specific signatures (i.e. oligonucleotide sequences that are present in all target sequences and absent in all near neighbors) and (iii) computational scale, determined by the number and length of target and non-target sequences. The lack of algorithms able to confront these growing challenges makes the design of many group-specific assays intractable.
Variation in the length and overlap of available sequences impedes assay design. Short sequences reduce the regions available for assays, and the lack of overlap (or only partial overlap) forces the omission of data that may contain informative biological variation. For example, if 1200 bp sequences were required as input for assay design, half of the 16S rRNA gene sequences in the Ribosomal Database Project (RDP) would be omitted. About 52% of sequences in the RDP (release 10, 5 April 2011) are less than 1200 bases (the full-length gene is 1400–1500 bp), and the sequences represent different regions of the 16S rRNA gene. Variation in sequence length also impedes efforts to determine the extent to which an assay or set of assays covers existing sequences. Current programs do not address the problem of variable length sequences.
The lack of target-specific signatures also restricts assay design. A signature sequence is unique to, and conserved within, the target group and thus confers assay specificity. With the exception of PRISE, computer programs for group-specific assay design require signature primers. This approach is attractive because it is computationally inexpensive, but it prevents the design of assays for groups that lack a unique signature. For such groups, specific assays can still be obtained by exploiting the specificity arising from thecombination of forward and reverse primers. That is, the primers can be individually non-specific, but group-specific as a pair ( Figure 1 ). This strategy was used to manually design a PCR assay that differentiatesBrucella abortus from the closely related B. suis , B. melitensis , and B. ovis. The assay targets genes that are present in all Brucella species but are uniquely arranged in B. abortus . Design programs that require a signature primer would not discover this primer pair. The PRISE program is capable of designing this type of assay but cannot accommodate design problems on a large computational scale (determined by the number and length of input sequences).
Figure 1.
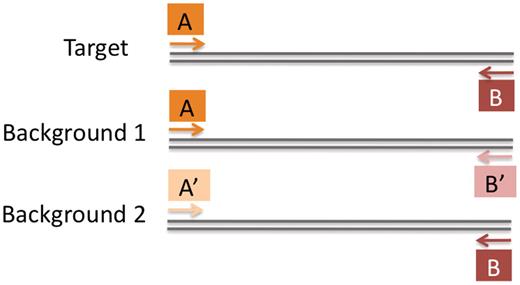
Non-signature primers can produce a target-specific assay. Primer sequences A and B occur in both the target and the background sequences, but only occur as a pair in target sequences. Instead of exploiting unique oligonucleotides (i.e. signatures), assays can exploit unique oligonucleotide combinations .
To address these limitations, we developed two algorithms: SeqStrap and ProSig. SeqStrap enables use of partially overlapping sequences that might otherwise be discarded, while ProSig performs assay design for different assay formats (e.g. PCR, TaqMan PCR and other probe-based PCR assays). Here, we demonstrate the sequential application of these algorithms for design of quantitative PCR (qPCR) assays specific for Acidobacteria Group 1. This group was chosen because it is ecologically important and technically challenging to target. TheAcidobacteria are of ecological interest due to their high abundance in soils, ∼20% of the total bacterial community , along with their response to pH, carbon, soil management and elevated atmospheric carbon dioxide . The phylum contains 26 rRNA gene sequence similarity groups, labeled Groups 1 to 26 . Acidobacteria Group 1 is of particular interest because it contributes to cellulose degradation and responds to ecosystem exposure to elevated atmospheric CO 2 , a factor in global warming.
The diversity of Acidobacteria Group 1 illustrates the scope of the assay design problem. There is little formal taxonomic structure within Group 1. Formal structure (i.e. the elucidation of taxonomically well-defined Orders, Families and Genera) requires analyses of representative bacterial cultures. These organisms are difficult to culture and there are only 5-sequenced genomes (S. Lucas, A. Copeland, A. Lapidus, J.-F. Cheng, L. Goodwin, S. Pitluck, H. Teshima, J. C. Detter, C. Han, R. Tapia et al. , unpublished results; S. Lucas, A. Copeland, A. Lapidus, J.-F. Cheng, L. Goodwin, S. Pitluck, A. Zeytun, J.C. Detter, C. Han, R. Tapia, et al. , unpublished results; S. Lucas, A. Copeland, A. Lapidus, J.-F. Cheng, L. Goodwin, S. Pitluck, A. Zeytun, J.C. Detter, C. Han, R. Tapia et al. , unpublished results) and 10 formally described cultured species in Group 1. Consequently, diversity within the Group is known mainly from rRNA gene sequences amplified from environmental samples. Among the 1339 nearly full-length (≥1200 bp) 16S rRNA gene sequences representing AcidobacteriaGroup 1 in the RDP , the most divergent sequences are ∼85% similar . Using a complete linkage clustering algorithm and thresholds corresponding to 95, 90 and 85% sequence similarity, we found the number of clusters formed by the 1339 sequences at each threshold was 132, 10 and 1, respectively. In taxonomic terms, these clusters provide a lower bound estimate of the number of genera, families and orders, illustrating the extensive breadth of diversity within the group. Many failed attempts have been made over the past decade to design PCR assays for Acidobacteria Group 1. Here, we demonstrate the success of the SeqStrap and ProSig programs in solving this complex design problem.
Acidobacteria 16S rRNA gene sequences were obtained from the RDP release 10 ( 33 ) on 21 August 2010. The sequence lengths ranged from 262 to 1512 bp for the Group 1 Acidobacteria , and from 235 to 1697 bp for the non-Group 1 Acidobacteria .
SeqStrap and ProSig were run on a 376-core, 2.5 GHz Intel Xeon-based computer cluster with a gigabit Ethernet network. SeqStrap requires a CPU that supports Streaming SIMD Extensions (SSE) version 4.1 (or higher) to compute the Smith–Waterman alignment of a single query sequence against four subject sequences in parallel using 32-bit alignment scores.
The two algorithms are implemented in C++ and compiled with gcc to run on the Linux operating system. The MPI software library is used to parallelize the calculation on a cluster of Linux computers. Each program is invoked independently via the command line or cluster batch scheduling software. Both ProSig and SeqStrap are publicly available as open source software (GNU public license version 2) and can be downloaded from http://public.lanl.gov/jgans .
SeqStrap is a computationally intensive algorithm that performs iterative, pair-wise sequence extrapolation as a preprocessing step before assay design. The algorithm, outlined in Figure 2 , iteratively extends each partial target sequence by adding the overhanging sequence from the most similar target sequence (illustrated in Figure 3). The most similar sequences must have an alignment score greater than a preset threshold and must be the highest scoring pair-wise alignment found by a search against all other target sequences. SeqStrap uses a conservative threshold score of 50 (see the scoring scheme described in the legend of Figure 2 ).
Figure 2.

Pseudocode description of the SeqStrap algorithm. Sequence alignments are computed using Smith–Waterman dynamic programming with the following scores: 2 for a match, −3 for a mismatch, −5 for gap existence and −2 for gap extension. No penalty is assigned for overhanging ends. When multiple sequence alignments produce the same highest score, the alignment with the largest amount of overhanging sequence is selected. When the amount of overhanging sequence is also the same, the order of the sequences in the input file is used to break the tie (to insure reproducible results). Examples of overhanging sequence are shown inFigure 3 . Two heuristic parameters are required: a score threshold and a maximum sequence length. The score threshold prevents spurious matches from creating sequence chimeras. Requiring a maximum sequence length was empirically found to prevent infinite extrapolation loops, e.g. terminal mismatches leading to an endless cycle of sequence A extrapolating sequence B, B extrapolating sequence C, and C extrapolating sequence A. All extrapolations used a score threshold of 50 and a maximum sequence length of 1500 bp. Because the maximum length criterion is applied before extrapolation, it is possible to produce extrapolated sequences that exceed the maximum sequence length.
Figure 3.

There are three possible ways to extrapolate a query sequence: ( a ) add the 3′-overhanging sequence from the best match, ( b ) add the 5′-overhanging sequence from the best match, and ( c ) add both the 5′- and 3′-overhanging sequence from the best match.
To reduce the number of unproductive sequence comparisons (i.e. alignments that do not contribute to sequence extrapolation), sequences are sorted by length in ascending order at the beginning of each iteration, and the shortest sequences are extrapolated first. Overlap alignments between pairs of sequences are computed with Smith–Waterman dynamic programming. Because only the alignment score and the coordinates of overhanging sequence are needed (as opposed to a detailed pair-wise alignment), SeqStrap uses a linear-space variant of the Smith–Waterman dynamic programming that only stores two rows in the dynamic programming matrix. This avoids the potential storage limitation in aligning longer (>10 5 bp length) nucleic acid sequences (the dynamic programming matrix size is proportional to the product of the aligned sequence lengths).
To reduce the computational burden of extrapolation with large numbers of sequences, SeqStrap exploits two levels of computer parallelism to compute the independent pair-wise alignments. At the lowest level, alignments between the query sequence (to be extrapolated) and the subject sequences (sources of extrapolated sequence) are computed in parallel. By using the 128-bit SSE available on modern Intel and AMD CPUs, four independent alignments (using 32-bit scores) can be computed in parallel. At the highest level, all sequences are uniformly distributed between available CPU cores in a cluster computer using the MPI parallel toolkit. For each query sequence, every core computes the alignments between the query and the locally stored subject sequences.
Even with this parallel implementation, 1 week on a 376-core cluster represents a significant investment of computational resources. The expended computer time reflects our choice of problem (16S rRNA gene sequences, one of the most abundant sequence types in public databases) and the desire for highly accurate, overlap sequence alignments (using Smith–Waterman dynamic programming). The required running time of the extrapolation algorithm is difficult to predict a priori and depends on several factors, including number of sequences, distribution of sequence lengths, pair-wise similarities between sequences and cluster hardware. However, a significant speed up in algorithm performance could be obtained by replacing the Smith–Waterman alignment algorithm with a faster (likely heuristic), sequence overlap alignment algorithm. The open source implementation of the SeqStrap algorithm can serve as a benchmark for future algorithmic improvements that trade alignment accuracy for alignment speed.
ProSig designs PCR, TaqMan PCR and probe-based group-specific assays by large scale assay enumeration followed by subtraction of enumerated assays that potentially cross-react with non-target sequences ( Figure 4 ). A greedy set-coverage solver then identifies the minimal sets of assays required to detect all targets. Multiple sequence alignments and signature oligonucleotides are not used in the design process. Large numbers of target sequences (ranging in size from genes to bacterial genomes) can be processed. In addition, because each forward and reverse primer will participate in multiple assays, we only perform computationally expensive, pair-wise sequence alignments once for each primer and template sequence. Finally, we exploit parallel computing to distribute both the required storage and sequence comparison calculations across a cluster of computers.
Figure 4.

Pseudocode description of the greedy PCR-assay enumeration algorithm. Core sequence refers to the original, non-extrapolated sequence. Primer and assay enumeration used the target parameters listed in Table 1 , while the removal of assays used the non-target parameters listed in Table 1 .
Assay enumeration is computationally demanding, but feasible with a mid-sized cluster (i.e. hundreds of cores). When enumerating assays from a selected target sequence, all valid forward and reverse primers from the core (i.e. non-extrapolated) region of the selected targets are enumerated. Valid primers are defined by user-specified constraints in length, primer-template melting temperature, primer-hairpin melting temperature and other common guidelines for primer design ( Table 1). Melting temperatures are calculated with a standard nearest-neighbor thermodynamic model that provides both melting temperature and free energy change given a pair-wise sequence alignment that can include mismatches and gaps.
Table 1.
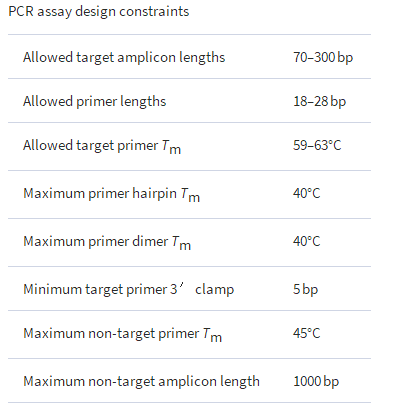
The constraints used to (a) enumerate PCR primers and assays from target sequences and (b) subtract PCR assays that matched non-target sequences. An assay matched a non-target sequence if both primers bound in the correct orientation, with melting temperatures ( Tm ) ≥ 45°C, and produced an amplicon ≤1000 bp. All melting temperatures were computed by a thermodynamic alignment between primer and template, using the nearest neighbor parameters of SantaLuci, and assuming a primer concentration of 9 × 10 −7 M and a salt concentration of 0.05 M. Target match criteria are more stringent that non-target match criteria.
The potential number of forward and reverse primer candidates is of order 2( nL ), where L is the size of the core target sequence (e.g. ∼1000 bp for 16S rRNA gene targets), and n is the number of allowed primer lengths (typically less than 10 different lengths ranging from 20 to 30 bp). While each primer can potentially participate in multiple PCR assays, the computationally expensive primer-template and primer-hairpin melting temperatures are only calculated once for each primer. When computing the primer-template melting temperature, only the perfect match is considered. Combining the enumerated primers, the number of possible PCR assays is order ( ΔAn 2 L ), where ΔA is the range of allowed amplicon sizes (i.e. largest amplicon size—smallest amplicon size; typically <200 bp). The calculation of the primer-dimer melting temperature for the forward and reverse primers must be computed for every assay. This calculation dominates the enumeration of valid assays from a single target sequence because the number of primer combinations is far greater than the number of primers. In practice, the number of melting temperature calculations is less than order ( ΔAn 2 L ), because only a fraction of possible primers satisfy the constraints shown in Table 1 , but a general computational framework is still needed that can perform ∼10 7 –10 10 independent melting temperature calculations per target sequence. The ProSig program provides this framework by distributing the melting temperature calculations in parallel on multiple CPUs in a computer cluster.
The computational cost of performing PCR-assay subtraction (i.e. matching and removing assays that potentially cross-react with non-target sequences) is of order ( 2nL ), which is far less demanding than the assay enumeration step . The melting temperature is calculated for each primer bound to each non-target sequence. Since primer binding can potentially occur anywhere in a non-target sequence, melting temperature calculations are initiated for all non-target sequence subregions that share at least six consecutive bases of perfect complementarity with a given primer (identified by a hash table lookup). All non-target sequence binding sites with melting temperatures above the user-defined threshold are stored for each primer. Assays that could conceivably produce amplicons with non-target sequences are rejected. Assay rejection occurs if two primer binding sites occur in the correct orientation, within the allowed non-target distance, and with melting temperatures above the non-targetTm threshold ( Table 1 ). Also, at least one of the primers must have one or more 3′ terminal bases that are perfectly matching to the non-target sequence to be suitable for polymerase extension. If both primers have 3′ terminal mismatches to the non-target sequence, then the assay is not rejected. All possible primer pair combinations are tested (i.e. forward/reverse, forward/forward and reverse/reverse). As with the enumeration step, the assay subtraction step is performed in parallel for each non-target sequence. Only those assays that survive the subtraction process for the i -th non-target sequence are searched against the i +1 non-target sequence.
When searching candidate assay signatures against multiple non-target sequences, we sorted the non-target genomes by degree of similarity to the target genome (most similar first) in order to remove the largest number of candidate signatures as early in the calculation as possible. Because candidate signatures that match a non-target sequence are removed from further consideration, this strategy reduces the number of signature searches that must be performed for subsequent non-target sequences. For the purpose of prioritizing non-target genomes for signature subtraction, genome similarity is defined by the magnitude of the difference between the normalized dinucleotide composition vectors for each genome [similar to ( 56 ), but using vectors of raw dinucleotide counts normalized to length one].
After assay subtraction, all remaining assays are predicted to be specific to one or more target sequences within the group. Identification of the smallest number of assays required to amplify all target sequences in the group is a standard set coverage problem ( 11 ). We used a standard greedy heuristic solution ( 57 ); after every iteration of enumeration and subtraction for a selected target sequence, the algorithm stores the assay(s) that amplify the largest number of previously unamplified target sequences. Target sequences amplified by previously stored assays are ignored. Not every target sequence is guaranteed to yield a specific assay (e.g. if the same sequence appears in both the target and non-target categories, then none of the enumerated assays will survive non-target subtraction).
The algorithm uses a process loop that consists of (i) selecting a previously unamplified target sequence, (ii) enumerating valid assays, (iii) subtracting assays that match non-target sequences, and (iv) finding the assay (or assays) that amplifies the largest number of previously unamplified targets. This process loop continues until all targets have been selected or amplified (or the process is manually terminated).
This approach fails in easily recognized modes when challenged with mislabeled sequences. If a target sequence is accidentally included in the non-target category, it will be ‘first in line’ for assay subtraction by virtue of its high similarity to other target sequences, and will quickly eliminate the majority of assay candidates. If a non-target sequence is accidentally included in the target category, its assays will be subtracted by other non-target sequences. Any assays that do survive (due to unique features in the mislabeled sequence) will be unlikely to detect other correctly labeled target sequences, and the set coverage algorithm will require a separate assay just to detect the mislabeled sequence. If a sequence that is dissimilar to both target and non-target sequences is included in the target category, it will generate a large number of assays that survive the non-target subtraction step but only detect the dissimilar sequence. Sequences that are dissimilar to both target and non-target sequences and are included in the non-target category will not affect the output (since they will not eliminate any assays during the subtraction step).
Assays were performed with Biorad iQ SyBr Green Supermix and three primer sets designed for specific detection of Acidobacteria Group 1 (Table 2 ). Primers were obtained from Invitrogen. Each 25 µl qPCR assay contained primers at 0.2 µM. Cycling conditions were as follows: 94°C for 5.0 min; 40 cycles of 94°C for 15 s, 65°C for 30 s; a melt curve of 91 cycles, 30 s each, ramping 0.5°C per cycle from 50.0 to 95.0°C; 4.0°C storage. Standard curves for primer sets AcidoG1_8.1 and AcidoG1_8.17 were generated with purified, genomic DNA from Acidobacterium capsulatum ATCC 51196 (hereafter A. capsulatum ) . A standard curve was not obtained for primer set AcidoG1_8.2 because it does not amplify A. capsulatum . Assays were applied to soil DNA samples from a field experiment in Rhinelander, Wisconsin, described in.
Table 2.
PCR primer pairs for the specific detection of Acidobacteria Group 1 16S rRNA gene sequences
Amplicons from triplicate qPCR reactions for each of three replicate soil DNA samples were pooled (i.e. n = 9 reactions), cloned and sequenced to evaluate assay specificity. Amplicons were purified (Qiagen QIAquick PCR Purification Kit) prior to cloning (Invitrogen TOPO TA Cloning Kit). For each qPCR primer set, 192 clones were picked and the cloned 16S rRNA gene fragments were bi-directionally sequenced with M13 primers.
Assembled sequences were visually inspected in Sequencher v4.7 (Ann Arbor, MI) to confirm the sequences were full length, as indicated by the presence of forward and reverse primer sites. A total of 149, 158 and 163 useable sequences were obtained for primer sets AcidoG1_8.1, AcidoG1_8.2, AcidoG1_8.17, respectively. Sequences were aligned in SILVA. Aligned sequences were compiled in a single database in ARB.
Phylogenetic placement of the amplicon sequences from qPCR assays was determined in two ways. First, the sequences were classified by an automated classifier in the RDP. Secondly, the sequences were added by quick Parsimony to a phylogenetic guide tree in ARB. The guide tree was generated with 145 nearly full-length (ranging from ∼1000 to 1500 bp) reference sequences representing AcidobacteriaGroups 1, 3, 4, 5, 6, 7, and 8. Archangium gephyra (β-Proteobacteria, AB218222), Solimonas soli (γ-Proteobacteria, EF067861), Chitinibacter tainanensis (β-Proteobacteria, AY264287) and Leeia oryzae (β-Proteobacteria, DQ280369) were used as additional out groups. The maximum likelihood algorithm (RAxML) in ARB was used to generate the guide tree with a 70% base frequency filter generated with Acidobacteria reference sequences representing Groups 1, 3, 4, 5, 6, 7 and 8. Amplicon sequences from the Acidobacteria Group 1 assays were added to the guide tree using the ARB parsimony algorithm, since the amplicons were short and represented different regions of the 16S rRNA gene.
The fold-change in relative abundance of groups targeted by qPCR assays was calculated as follows: (amplification efficiency) [(mean CT ambient)–(mean CT elevated CO2)] . In this calculation, the amplification efficiency term was computed from the slope of the PCR standard curve generated withA. capsulatum using e( − 1/slope) . The amplification efficiency was 1.85 and 1.94 for assays AcidoG1_8.1 and AcidoG1_8.17. The amplification efficiency of the third assay, AcidoG1_8.2, was not determined because the assay does not amplify A. capsulatum (the only available genomic DNA for the test) . Therefore, the amplification efficiency of the first assay, AcidoG1_8.1, was used to approximate the fold-change calculations for AcidoG1_8.2. Similar results were obtained when using the amplification efficiency of AcidoG1_8.17 as a substitute value.
The 470 amplicon sequences from the three assays are included in both the supplementary online material and the ProSig software package.
To maximize the number of sequence inputs for PCR-assay design, 8430 Acidobacteria Group 1 sequences and 25254 other Acidobacteria 16S rRNA gene sequences ranging from 235 to 1697 bp in length were separately processed with SeqStrap. Sequence extrapolation took about 1 week on a 376-core cluster. Extrapolation increased the length of 96% of the Acidobacteria Group1 sequences and 94% of the otherAcidobacteria sequences. Extrapolation increased the average length of target sequences from 759 to 1469 bp and the average length of non-target sequences from 826 to 1512 bp. Sequence extrapolation occurs only between a sequence and the single most similar matching sequence, identified by the largest pair-wise sequence alignment score (above a set threshold) among the pool of sequences to be extrapolated. The default alignment-scoring scheme and the minimum allowed score for sequence extrapolation ( Figure 2 ) allows extrapolation between pairs of sequences with (gap free) sequence identities >60% (in the long sequence alignment limit). This constraint reduces, but does not eliminate, the generation of chimeric artifacts by extrapolation. However, since our group-specific assay design process works by enumerating candidate assays only from the unextrapolatedportion of target sequences, chimera-specific assays will not arise. It is still possible for chimeric sequences to cause a reduction in predicted assay coverage (i.e. the number of target sequences detected by a given assay). To our knowledge, the SeqStrap algorithm is a unique approach that overcomes the challenge of fragmentary data in sequence databases and, thus, maximizes the data that can be exploited for assay design. In our pipeline, the extrapolated sequences are used during the design process to assess the extent to which assays can amplify existing sequences.
ProSig was used to enumerate target-specific assays. The AcidobacteriaGroup 1 sequences did not have a specific signature that could be used for a PCR assay. The lack of a signature and the large number of sequences made this target group a suitable test of ProSig’s design capability. The cumulative fraction of Acidobacteria Group 1 sequences covered by the assays was monitored in real-time by computationally searching the assays against the sequences extrapolated by SeqStrap, and for comparison, to the original, unmodified (i.e. unextrapolated) sequences. The first four assays were predicted to cover ∼50% of the extrapolated Acidobacteria Group 1 sequences, whereas about 15 assays were predicted to cover 50% of the unextrapolated sequences ( Figure 4). This illustrates one of the challenges posed by use of unextrapolated sequences that vary substantially in length and overlap. After the first 40 assays (providing ∼95% predicted cumulative coverage), the predicted coverage of Acidobacteria Group 1 extrapolated sequences plateaued ( Figure 5 ). We terminated the ProSig run after generating 83 assays because further improvements in predicted coverage were marginal ( Figure 5 ). The 83 assays were predicted to collectively cover ∼98% of the Acidobacteria Group 1 sequences and required ∼48 h to compute on our 376-core cluster. The forward primers for the assays collectively targeted about 24 distinct locations in the 16S rRNA gene. The reverse primers targeted about 19 locations. The inability to detect the entire target group with a single assay emphasizes the difficulty of the assay design problem. Similar results were obtained using theActinomycetales as a target group (data not shown), demonstrating thatAcidobacteria Group 1 is not unique in posing a design challenge, and emphasizing the need for a robust, flexible design platform like ProSig. The ability of ProSig to predict a minimal set of assays to coverAcidobacteria Group 1 illustrates its flexibility and value.
Figure 5.
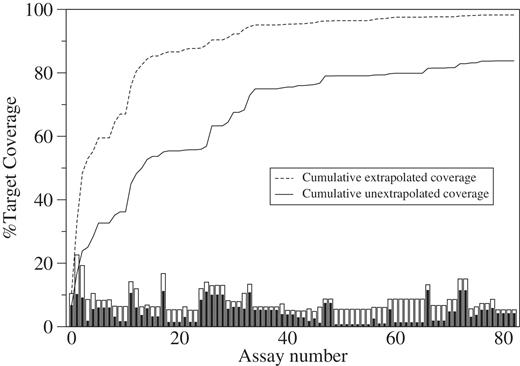
Cumulative coverage of 8430 Acidobacteria group 1 16S rRNA gene sequences by 83 target-specific PCR assays. Coverage is the percentage of the extrapolated or unextrapolated (unmodified) target sequences amplified ( in silico ). The bar height represents the extrapolated (white bars) or unextrapolated (black bars) target coverage of individual assays. The dashed and solid lines represent the cumulative extrapolated and unextrapolated target coverage, respectively. PCR assays are plotted along the x -axis in the order of discovery. The maximum cumulative coverage is 98% for the extrapolated targets and only 84% for the unextrapolated targets. The coverage of individual assays ranges from a high of 23% to a low of 5% for the extrapolated targets, and from a high of 11% to a low of 1% for the unextrapolated targets.
Three Acidobacteria Group 1 assays were chosen for experimental testing. To find the optimum subset of three assays predicted to collectively amplify the largest fraction of extrapolated target sequences, a brute force search was performed with all 91 881 possible three-assay combinations from the pool of 83 target-specific assays [i.e. the number of three assay combinations = 83!/(3! × 80!)]. The Perl script used to perform this search is included with the ProSig software. It is not possible to determine an optimal subset of assays simply by examining Figure 5 , because each assay may detect overlapping subsets of the target taxa. In addition, because the number of possible assay combinations grows rapidly, the brute force solution to the set-coverage problem is only practical for small combinations (i.e. less than 5). The optimum combination of three assays was AcidoG1_8.1, AcidoG1_8.17 and AcidoG1_8.2 ( Table 2 ). Using E. coli numbering, the assays, respectively, target the following three regions of the 16S rRNA gene: basepair 906 to 1071 (between variable regions V5 and V6), bp 373 to 607 (encompassing V4) and basepair 596 to 813 (encompassing V3), respectively. The three assays were predicted to collectively cover 42% of the extrapolated set of 8430 Acidobacteria Group 1 sequences. The predicted clade coverage of the three assays is shown inSupplementary Figure S1 . None of the primers in the three assays were ‘signature’ primers, illustrating the unique ability of ProSig to design assays for target groups that lack group-specific signatures.
The specificity of the assays was confirmed by sequencing PCR amplicons derived from soil samples. Soil is an excellent test-bed for assay specificity because the hyperdiversity of species in soil microbial communities increases the opportunity for PCR primer cross-reactivity, and thus, loss of specificity. The RDP classifier identified nearly all of the 149 (for AcidoG1_8.1), 158 (for AcidoG1_8.2) and 163 (for AcidoG1_8.17) sequenced amplicons from each assay asAcidobacteria Group 1. The exceptions were 16 sequences from AcidoG1_8.1, which could not be classified with a confidence score >70%. This result was not surprising because the region of the 16S rRNA gene targeted by AcidoG1_8.1 does not provide good resolving power for the RDP classifier. More reliable classification was obtained by aligning sequences using a 16S rRNA gene-specific alignment strategy followed by reconstruction of a phylogenetic tree. When placed in a phylogenetic tree, all of the amplicon sequences fell within Acidobacteria Group 1 ( Figure 6 ), confirming the predicted specificity of the assays.
Figure 6.

Maximum-likelihood tree of Acidobacteria 16S rRNA gene sequences. Groups identified as 1, 3, 4, 5, 6, 7 and 8 are indicated to the right of the group. Sequences obtained from soil by PCR amplification with Acidobacteria Group 1 primer sets are indicated by color. The total number of sequences in each cluster (including both sequenced amplicons and reference sequences) is labeled at the cluster node. The number of sequenced amplicons for each respective PCR assay is shown in parentheses adjacent to the colored wedge. Wedges are colored to show the approximate fraction of amplicon from each PCR assay in the cluster (blue for 8.1, gold for 8.2 and purple for 8.17). Clusters containing reference sequences from cultured isolates are indicated. The scale bar indicates 0.10 changes per nucleotide.
The three group-specific assays were used to evaluate the response ofAcidobacteria Group 1 in a field experiment focused on terrestrial ecosystem responses to a decade of elevated atmospheric CO 2. The field experiment included three replicate field plots under ambient or elevated atmospheric CO 2 conditions. A composite soil sample was obtained from each plot, yielding six samples total. Exploratory Sanger-based, 16S rRNA gene surveys (about 270 sequences each) of the six samples showed a 2-fold decrease in the relative abundance of sequences classified as Acidobacteria Group 1 in plots under elevated CO2 ( Figure 7 ), but the difference was not statistically significant by a pair-wise t -test. Subsequently, Pyrotag-based, single subunit amplicon libraries (100-fold larger that the Sanger surveys) of the same samples showed a significant ( P = 0.017), 3-fold decrease inAcidobacteria Group 1 under elevated CO 2 . To investigate further, the three qPCR assays, AcidoG1_8.1, AcidoG1_8.17 and AcidoG1_8.2 were applied to the same samples. Although the three assays do not detect all members of Acidobacteria Group 1, we expected the assays would cover a sufficient number of species to capture the Group 1 responses observed in the 16S rRNA gene surveys. With AcidoG1_8.1, there was no difference in abundance between ambient and elevated CO 2 samples. In contrast, ∼5-fold decreases in abundance under elevated CO 2 were measured with AcidoG1_8.17 and AcidoG1_8.2, and the differences were significant ( P = 0.0083, P = 0.0023, respectively). The results demonstrate the value of the assays in validating and further characterizing results obtained from broader survey techniques. The ability to identify responsive target groups from survey data and rapidly follow-up by designing and applying group-specific assays is an important capability in microbial ecology.
Figure 7.
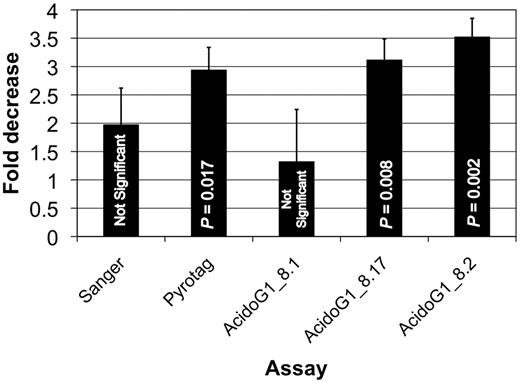
Decrease in the relative abundance of Acidobacteria Group 1 in a field experiment measured by different techniques applied to six soil DNA samples from control (ambient atmospheric CO2 ) and treated (elevated atmospheric CO 2 ) plots. The Sanger estimate was obtained from surveys of about 300 16S rRNA gene sequences from each soil DNA sample. The Pyrotag estimate was obtained from surveys of about 50 000 16S rRNA gene sequences from each sample. The variation in the estimates is presumed to arise from the increasing quality of the assays (from left to right) and the specific members of Acidobacteria Group 1 sampled by each technique.
We are aware of only one other program that uses an approach comparable to ProSig. Like ProSig, the PRISE program permits discovery of specific PCR assays composed of non-signature primers, but the program has several limitations that severely constrain its capability. The program performs a limited enumeration of valid, target-sensitive PCR assays. Screening the enumerated assays against all non-target sequences then identifies target-specific assays. However, this program requires a greedy enumeration, ignoring primers that do not match a significant fraction of the target sequences. When this constraint is omitted, the program exhausts the available memory, even for small problem sizes (i.e. a small number of target and query sequences). Thus, this program is suitable only for problems in which most of the targets can be detected by an assay.
We developed a computational methodology that exploits available nucleic acid sequence information at an unprecedented scale and level of complexity to develop robust PCR-based detection assays. This methodology enables the design of group-specific assays for a wide range of applications in microbial ecology, public health and agricultural safety. The last 20 years have witnessed an exponential growth in the number of available nucleic acid sequences. Improved computational tools that can scale with the growth of sequence information are still needed. The SeqStrap and ProSig help address this need. The software has been released as open source to facilitate future improvements in parallel scalability and novel assay design algorithms.