

发布者:primerbank 时间:2017-08-30 浏览量:2819
Andreas Untergasser, Ioana Cutcutache, Triinu Koressaar ,Jian Ye ,Brant C. Faircloth,Maido Remm ,Steven G. Rozen,
Nucleic Acids Research, Volume 40, Issue 15, 1 August 2012, Pages e115,
https://doi.org/10.1093/nar/gks596
Published:21 June 2012


The design of polymerase chain reaction (PCR) primers, like the laboratory technique of PCR itself, is ubiquitous and diverse. PCR is used for creation of templates for Sanger and next-generation sequencing, detection of the presence or absence of particular deoxyribonucleic acid (DNA) sequences, assessment of the length of simple sequence repeats, creation of constructs for genetic engineering, amplification of complementary DNA to detect splice forms and, using real-time PCR, measuring transcript abundance or DNA copy number. The diversity of PCR applications requires corresponding flexibility in programs for PCR primer design.
The most cited software for primer design in recent years includes PrimerSelect (1), Primer Express (2), Primer Premier (http://www.premierbiosoft.com/primerdesign/index.html), the ‘OLIGO’ software series [most recently OLIGO 7 (3,4)] and Primer3 (5). In addition, Visual oligonucleotide modeling platform (OMP) (6) is noteworthy for its detailed physical modeling of many aspects of PCR primer design, including the effects of modified nucleic acids and PCR buffer additives, as well as a partial ability to simulate PCR reactions.
Primer3, the subject of this article, is a popular group of programs, programming libraries and web interfaces that assist researchers with PCR primer design (Figure 1). The initial version of Primer3, which was released over a decade ago (5), was designed to select primers and hybridization oligonucleotides (‘oligos’) for the detection of a DNA sequence (i.e. for the design of assays for ‘STSs’—sequence tagged sites) and for design of PCR assays for microsatellite genotyping. Given a template DNA sequence, the initial version of Primer3 evaluated acceptable primers and primer pairs and then returned the pairs with the lowest penalties. Primer3 consisted of a command-line program that could be integrated into bioinformatic pipelines, whereas a basic web interface, Primer3web (http://primer3.wi.mit.edu/), allowed the use of Primer3 through the internet.
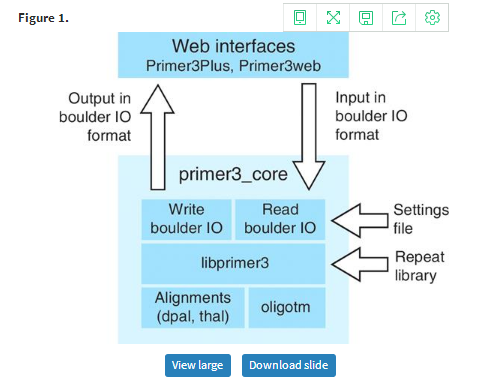
Organization of Primer3. The web interface communicates with primer3_core using the boulder IO format, as described in the text and Figure 4. The primer3_core main program uses a library for reading boulder-IO arguments from the input stream or from a settings file and then calls choose_primers() in the libprimer3 library. Libprimer3 can also read a repeat library, i.e. a library of highly prevalent repeats that primer pairs should avoid amplifying. For example, for the human genome these repeats include Alus, LINEs, endogenous retroviruses and simple sequence repeats (microsatellites, see RepBase, ftp://ftp.ncbi.nih.gov/repository/repbase/). Libprimer3 calls several other C/C++ libraries that are part of Primer3. The library for thermodynamic alignments for secondary structures, etc. is called thal. The library for the old-style alignments is dpal. The library for calculating oligo melting temperatures is oligotm. The primer3_core main function returns its results using a library to write boulder-IO format.
Since its initial release, Primer3 has become extremely popular. It was downloaded >20 000 times during 2011, and >7000 manuscripts reference the original publication (Google Scholar). Primer3 likely owes its popularity to several factors that include the availability of a relatively easy-to-use web service, robust engineering, open access to the program source code, suitability for use in high-throughput pipelines for genome-scale research and the ease with which Primer3 can be incorporated into or interoperate with other software. Supplementary Table S1 lists >25 third-party software tools and web services that incorporate Primer3. Primer3’s popularity also likely stems from its ability to meet the needs of three groups of users: (i) bioinformaticians, who embed Primer3 within their web services, bioinformatics scripts or pipelines, (ii) expert users who select large numbers of primers and need tight control over the selection process and (iii) occasional users who simply want a convenient way to design a few primer pairs. Bioinformaticians usually rely on the Primer3 command-line interface and run Primer3 on a local machine. The other users, whether ‘expert’ or ‘occasional’, usually rely on Primer3’s web interfaces.
In light of the multiple uses of PCR, it is not feasible for a single program to meet every need. To overcome this limitation, we envisioned Primer3 as a ‘software component’ and have encouraged the integration of Primer3 into other software or web services (5). Indeed, Primer3 is unique in the degree to which it has been incorporated into programs and web services tailored to specific primer design needs: multiple software packages and web services have built on Primer3’s capabilities by organizing and enhancing Primer3’s inputs and outputs to meet specific objectives. We mention several examples in this article and list others in Supplementary Table S1. PRIMEGENS is both a stand-alone program and a web service for high-throughput primer design that offers several integrated functions, which include genome-wide specificity checking and automatic use of gene annotations (7). It also offers dedicated functionality for designing primers to amplify bisulfite-converted DNA (to assess methylation) (8) and for designing specific primers even if highly similar sequences are also present in the template DNA (9). ‘The PCR suite’ web service (10) integrates Primer3 functionality with GenBank annotations of gene and single-nucleotide polymorphism (SNP) locations and also supports tiling of PCR products across a region. Primer-BLAST is a web service that supports the selection of specific primers by considering opportunities for mispriming across an entire genome or transcriptome (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (11). The BatchPrimer3 web service (12) allows high-throughput primer design from a list of target sequences and also extends Primer3’s selection methods with a focus on SNP typing.
Since we first released Primer3, the fields of bioinformatics, genomics and molecular biology have expanded rapidly, and the amounts and types of sequence data that biologists collect and process are vastly greater and more diverse. In addition, the field has developed more accurate algorithms for predicting the hybridization behavior of nucleotide sequences. These changes have provided opportunities to improve Primer3’s functionality:
· The thermodynamic models available for predicting primer melting temperatures, binding and formation of primer dimers and secondary structure have been updated and refined (13–20). Primer3 now offers these up-to-date models, described in detail later.
· Whole-genome sequences are now available for many species. Thus, it is possible in principle to electronically estimate the likely specificity of candidate primer pairs, and one common approach is to search for and then avoid segments of the intended amplification target that are too similar to other sequences (6,7,21). Although we have not added a ‘search-and-avoid’ capability to Primer3 itself, we have enhanced Primer3, so that it can better support software that uses this approach. This support consists of new ways to precisely constrain primer location, for example, to constrain primers to locations with minimal similarity to other sequences. In addition, for reverse-transcriptase (RT) PCR, Primer3 can now require primers that span exon–exon junctions.
Finally, the spread of molecular techniques to all corners of biology brings an even wider variety of users to Primer3—from experienced bioinformaticians to high-school students designing and generating their first PCRs. Thus, there has been interest in augmenting Primer3 with interfaces targeted for specific design tasks and in further increasing the ease with which Primer3 can be integrated with other software. This has motivated several enhancements. These include the ability to require that primers not be used in multiple primer pairs, the ability to save and re-use argument settings in both the command line (primer3_core) and web interfaces, rationalization of the names of the arguments and a programming library interface for calling Primer3 functionality directly from other programs.
We have greatly expanded Primer3’s functionality since the original release. In this article, we detail the major features that improve the capabilities and accuracy of Primer3 for designing primers, simplify users’ interaction with Primer3 and simplify the integration of Primer3 with other software.
The most visible components of Primer3 are the web interfaces, Primer3Plus (http://primer3plus.com) (22) and Primer3web (http://primer3.wi.mit.edu) and the command-line primer-design program, primer3_core (Figure 1). Primer3_core is mostly useful to bioinformaticians with programming skills and is written with the goal of being computationally efficient, thoroughly testable and easily integrated with other software. Primer3Plus (Figures 2 and 3) and Primer3web offer interfaces more suitable to ‘expert’ or ‘occasional’ users. However, Primer3_core is the engine underlying all primer design operations, and most of the changes described in this article affect both the web interfaces and primer3_core, which we collectively refer to as Primer3.
Figure 2.

Input web page for Primer3Plus.
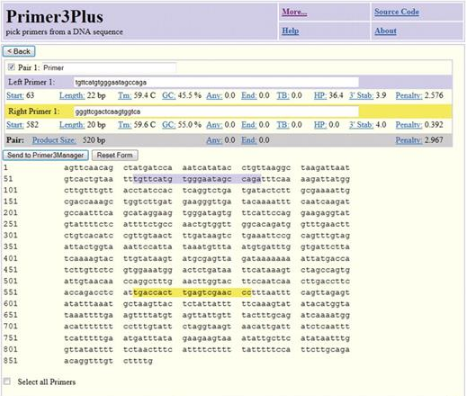
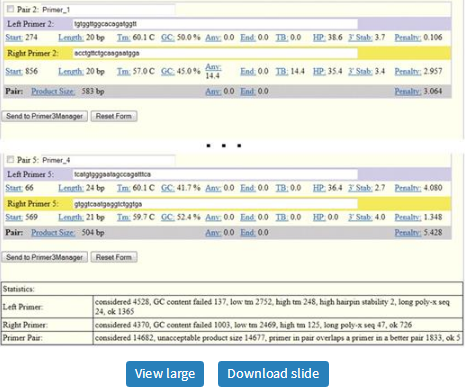
Example output web page from Primer3Plus. The first major block, labeled ‘Pair 1’, shows the template sequence with locations of the primers highlighted in blue and yellow and key information about the primers and primer pair. Subsequent blocks (‘Pair 2’,…, ‘Pair 5’) show information for alternative primer pairs.
Primer3 can carry out several kinds of design tasks (discussed later) and also check existing primer pairs for correctness. We focus mainly on discussing Primer3 in the context of designing primer pairs for amplifying a DNA template using PCR. To accomplish this task, Primer3 evaluates the primers and primer pairs according to various constraints and sorts acceptable pairs by a penalty function. It uses ‘branch and bound’ techniques (23) to reduce the search space while still generating the optimal primer pairs according to the penalty function and constraints.
Because the primer3_core program accepts >150 parameters and long template sequences—far too much information to be supplied on the command-line—it reads most of its inputs from flat text files. Specifically, primer3_core uses the ‘boulder IO’ (boulder input-output) format, in which each parameter name (termed a ‘tag’) and its corresponding value are joined by an '=' sign, with one tag and its value per line (Figure 4). A single input file can specify multiple primer design tasks: the inputs for individual design tasks are separated by lines consisting only of an '=' sign. Primer3_core also generates its output in boulder IO format (Figure 4B). This format is designed for integration with other software, including scripts and web services, on the assumption that end users will interact with the web interfaces.
Figure 4.
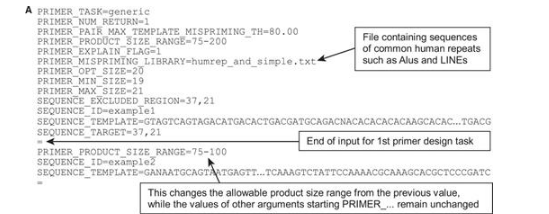
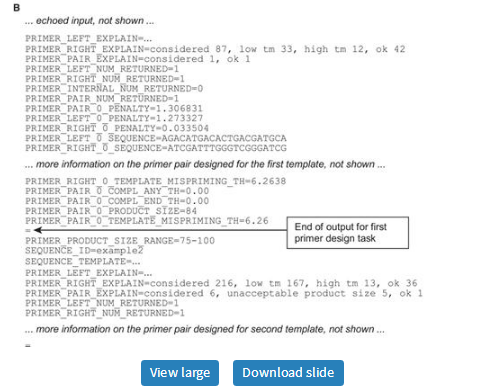
Examples of boulder IO (A) input to primer3_core and (B) output from primer3_core. Panel A shows two primer design tasks (SEQUENCE_ID’s example 1 and example 2) separated by a line consisting only of an = character. PRIMER_NUM_RETURN is 1 for both tasks, because the values of tags beginning with PRIMER_… persist between design tasks. However, the value of tags beginning with SEQUENCE_…, such as SEQUENCE_EXCLUDED_REGION = 37,21 in example 1, affect only the current design task. Panel B shows abbreviated output corresponding to panel A.
The most recent primer3_core release is available at http://sourceforge.net/projects/primer3/files/primer3/. The Primer3Plus web service is available at http://primer3plus.com/cgi-bin/dev/primer3plus.cgi. The most recent Primer3web code is available at http://sourceforge.net/projects/primer3/files/primer3-web/, and the Primer3web service is available at http://primer3.wi.mit.edu/.
Primer3_core consists of a main function and several C++ libraries (Figure 1), including libraries that calculate the melting temperatures of oligos, libraries that calculate the propensity of oligos to form hairpins or dimers or to hybridize or prime from unintended sites in the genome, libraries for reading and writing boulder-IO and the library libprimer3, which is a stand-alone C/C++ library that provides Primer3’s central primer-design capabilities. When called to select primers, libprimer3’s central function, choose_primers(), performs an exhaustive search for the ‘best’ legal primer pairs given the specified template sequence and other input arguments. ‘Legal’ primers or primer pairs are those that satisfy user-specified constraints, such as minimum and maximum melting temperatures for primers, minimum and maximum product size and so forth. Additional constraints govern possible locations of primers and primer pairs, as we discuss in more detail later. The notion of ‘best’ primers or primer pairs is operationally defined as minimizing a penalty function, as described in the documentation (http://primer3.sourceforge.net/primer3_manual.htm).
We have updated Primer3’s thermodynamic models for melting temperature calculations and have added thermodynamic models for estimating the propensity of primers to hybridize with other primers or to hybridize at unintended sites in the template; we have also added thermodynamic models for calculating the stability of potential hairpin structures within primers. All thermodynamic models now use up-to-date nearest-neighbor parameters and salt correction formulae.
Nearest-neighbor thermodynamic models offer the most accurate approach for predicting the energetic stability of DNA structures (19). In brief, these models estimate duplex stability by considering not only the effects of the number of hydrogen bonds linking base pairs but also the effects of the stacking of neighboring base pairs along the length of the duplex. For example, because of stacking, the stability of 5′-CT-3′ hybridized to 3′-GA-5′ is different from that of 5′-CA-3′ hybridized to 3′-GT-5′, even though the base pairings—C:G and T:A—are the same. To account for the effects of base-pair stacking, nearest-neighbor models capture the thermodynamic stability of overlapping neighboring base pairs rather than considering only single base pairs at a time. For example, nearest-neighbor models calculate the melting temperature of the oligo ACTGCG using the thermodynamic parameters of the overlapping neighboring bases that make up the sequence: AC, CT, TG, GC and CG. These models (and Primer3) also consider specific aspects of the PCR buffer composition—salt and deoxyribonucleotide triphosphate (dNTP) concentrations—that influence the stability of primer binding and potential secondary structures.
Using these thermodynamic models, Primer3 calculates the melting temperatures of oligos when hybridized to their intended targets and also predicts the strength of three types of secondary structures with possible mismatches: two types of bimolecular hybridization (‘ANY’ and ‘END’ interactions) and one type of unimolecular folding. Bimolecular ANY interactions involve the binding of an oligo (a primer or a hybridization oligo), anywhere within its sequence, to another DNA molecule (Figure 5A and B). Bimolecular END interactions involve the binding of the 3′-end of a primer to another single-stranded DNA molecule (Figure 5C and E). Some, but not all, END interactions are also ANY interactions, and vice versa, and the most stable ANY interaction is always at least as stable as the most stable END interaction. END interactions are most relevant to priming polymerases activity. Primer3 filters out primers with stable END interactions to avoid those prone to generating short ‘primer-dimers’ (Figure 5C). Primer3 also uses a thermodynamic model to assess the propensity of a primer or hybridization oligo to form unimolecular hairpin structures (Figure 5D). Because these can inhibit primer hybridization to template molecules and cause PCR failure (20), Primer3 filters out primers likely to form hairpins.
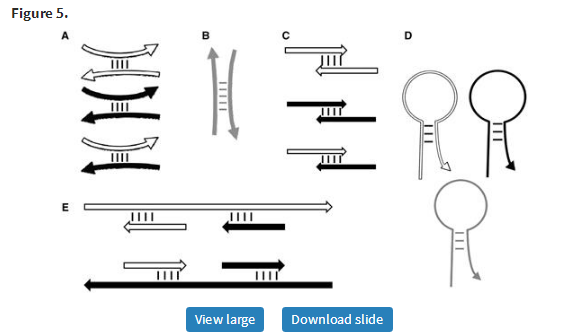
Primer3’s thermodynamic models for predicting the stability of secondary structures. (A–C) Interactions between primers. (D) Hairpin structures. (E) Undesirable binding of primers to template sequence. Short white and black arrows represent forward and reverse primers, respectively. Gray arrows represent hybridization oligos. Long white and black arrows represent forward and reverse template. Curved oligos (including primers) indicate ANY interactions (A, B and D), and straight oligos indicate END (i.e. 3′-anchored) interactions (C and E). Primer3_core and the Primer3 web interfaces now use thermodynamic models by default. The Primer3 manual (http://primer3.sourceforge.net/primer3_manual.htm) and Supplementary Figure S1 document the arguments governing temperature calculations for secondary structures.
Primer3 expresses the stability of ANY and END bimolecular interactions and of hairpin structures as melting temperatures—the temperatures at which there is a 50% possibility that a molecule is in a single-stranded form and a 50% possibility that a molecule has formed a duplex. By default, Primer3 requires these temperatures to be substantially lower than the minimum primer melting temperature.
The methods for calculating melting temperatures for ANY and END bimolecular interactions and of hairpin structures are extensions of those used to calculate melting temperatures for oligos when hybridized without mismatches to their intended target; in addition to considering the stability of neighboring base pairs, the secondary structure models also take into consideration the thermodynamic consequences of mismatched base pairs, gaps (physically, bulges) and loops. The relevant parameters are taken from the following: (13–20).
The Primer3 package also provides two stand-alone command-line programs: oligotm calculates oligo melting temperatures and ntthal (nucleotide thermodynamic alignment) predicts the propensity of oligos to form dimers or hairpins.
The availability of whole-genome sequences from many species (24) offers the possibility of using an organism’s genome sequence to improve primer-pair specificity. One approach to increasing PCR specificity is to search the intended target sequence against the rest of the genome and then restrict primers to regions within the target sequence that are minimally similar to other sequences in the genome. This approach can also be applied to primers for RT-PCR, by searching the intended target sequence against the entire transcriptome. This ‘search-and-avoid’ approach is used by GENOMEMASKER (21) and is one of the techniques used by Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/)(11). Furthermore, if almost the entire intended template is similar to another region of the genome, we might be satisfied if either the forward or reverse primer is in a region having minimal similarity to other regions of the genome. To flexibly support these situations, Primer3 now offers the argument SEQUENCE_PRIMER_PAIR_OK_REGION_LIST. This argument takes a list of region pairs and demands that the selected forward and reverse primers be in regions specified by at least one of the pairs in the list. For example, the list in Figure 6 specifies that there are three, equally acceptable alternatives for the locations of the forward and reverse primers:
· Forward primer in the 50 bp region starting at position 100 and reverse primer in the 50 bp region starting at position 300.
· Forward primer in the 50 bp region starting at position 400 and reverse primer anywhere.
· Forward primer anywhere and reverse primer in the 30 bp region starting at position 460.
An additional new argument in Primer3 supports a common requirement for RT-PCR, in which, to lessen the likelihood of amplifying genomic DNA or unspliced transcripts, it is often desirable to have one of the primers fall across the junction of two exons. To specify this, Primer3 offers the argument SEQUENCE_OVERLAP_JUNCTION_LIST. The value of this argument is a list of positions (e.g. of exon–exon junctions), and either the forward or the reverse primer must overlap one of these positions. By default, the minimum overlaps default to four bases at the 3′-end of the primer and seven bases at the 5′-end of the primer.
Figure 6.
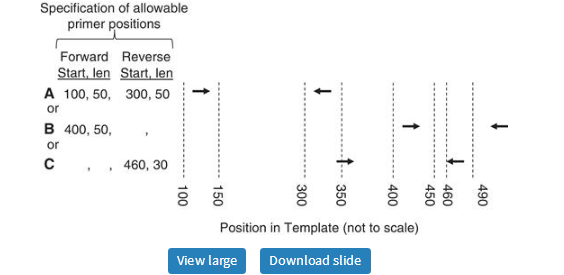
Constraining the positions of forward and reverse primers using SEQUENCE_PRIMER_PAIR_OK_REGION_LIST. Arrows represent primers. In this example, primer pairs A, B and C are all allowed. In the specification of allowable primer positions, blanks for ‘Start’ and ‘len’ indicate no constraint (example B, reverse primers, example C, forward primers).
Primer3 retains the other, previously available arguments for constraining the locations of primers and hybridization oligos. These include SEQUENCE_INCLUDED_REGION, SEQUENCE_EXCLUDED_REGION, SEQUENCE_TARGET and others. Please see the documentation (http://primer3.sourceforge.net/primer3_manual.htm) for details regarding the use of these arguments.
The current version of Primer3 remedies a common source of frustration for users of earlier versions: Primer3 often re-used the same forward or reverse primer in multiple primer pairs (Figure 7). In other words, the same primer was used several times in combination with a different mate. Thus, although primer pairs were unique, the primers within the pairs were not. Primer3 now allows the selection of primer pairs where each individual primer is used only once. In addition, one can guarantee that the multiple primer pairs returned by Primer3 are sufficiently different from each other by using the tag PRIMER_MIN_THREE_PRIME_DISTANCE. This tag ensures that the 3′-end of a forward (or reverse) primer has a minimum distance from the 3′-end of the forward (respectively, reverse) primer in any other primer pair (Figure 7).
Figure 7.
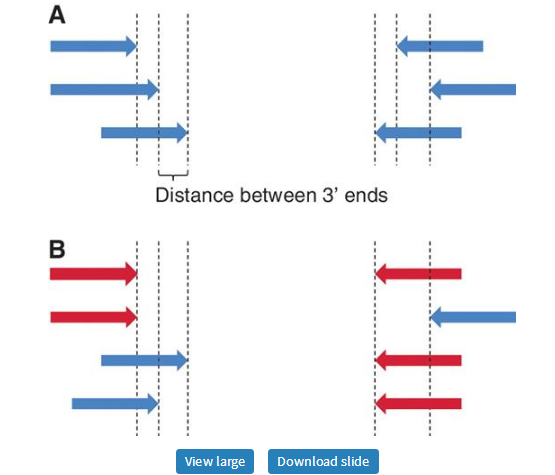
Unique and non-unique primers. (A) The 3′-end of each forward and reverse primer has a unique position; Primer3 can now ensure this and can ensure a minimum distance between the 3′-ends of all forward and/or all reverse primers. (B) Each primer pair is unique, but the red primers are used more than once.
For easy integration of primer3_core with other software, the boulder IO input tags must be straightforward to create, and the output tags must be straightforward to interpret by other programs or scripts. To this end, we have systematized tag names throughout the program. In particular, tags providing information specific to the current template sequence start with ‘SEQUENCE_’ (Figure 4A) and only have effect for a single primer design task; tags providing global information required for primer selection start with ‘PRIMER_’ and are active until they are over-ridden. We have also systematized the structure of the output tags to ease automatic parsing.
We have updated Primer3’s documentation (http://primer3.sourceforge.net/primer3_manual.htm) to explain each input and output tag in detail and to explain the details of the penalty calculations, which are governed by tags. The documentation also provides a table detailing the correspondences between old and new tag names.
Because the behavior of Primer3 is governed by >150 arguments, which often require tuning for specific tasks, we have added the ability to store and re-use argument values in ‘settings files’. These are text files containing a header followed by boulder-IO-formatted input that sets the values of ‘global’ tags (input tags prefixed by ‘PRIMER_’). Settings files offer a means to save and re-use argument settings developed for a particular application and a means for expert users to provide less-experienced users with parameter values suitable for their needs. Settings files can also be provided in publications as supplemental data to improve experimental reproducibility. For users of the web interfaces (Primer3Plus and Primer3web), settings files have the critical function of easing repetitive primer selections by obviating the need to manually change multiple parameters at the start of each design session. Users of the web interfaces can also download their current parameter values to a settings file and later, re-use these parameter values by uploading them back to the web interface or by using them on the command line with primer3_core.
To make Primer3 easier to use, we have re-factored the set of ‘tasks’ that Primer3 can carry out to include the following (Figure 8):
· generic: This task includes the standard design of primer pairs for PCR amplifying a DNA template (Figure 8A). When this task is specified, Primer3 can optionally design a hybridization oligo within the amplified region.
· check_primers: Check existing primers or primer pairs, even in the absence of a template sequence. Primer3 assesses whether the primer or primer pair satisfies the user specified constraints and also returns additional measures describing aspects of the primers and primer pairs (primer melting temperature, propensity to form secondary structure, product size, etc.)
· pick_sequencing_primers: Pick multiple unpaired primers for Sanger sequencing reads that will cover the template (Figure 8B). The spacing between successive forward primers and successive reverse primers is governed by arguments (described in the manual, http://primer3.sourceforge.net/primer3_manual.htm), but as the primers are designed to prime Sanger sequencing reactions, forward and reverse primers are not paired. This is different from selecting primer pairs to ‘tile’ a large region with PCR products for subsequent sequencing, which Primer3 does not offer [although the PCR Suite (10) layers this capability on top of Primer3.]
· pick_primer_list: Generate lists of legal forward and reverse primers without regard to their positions relative to each other (Figure 8C). This is approximately a re-implementation of the capability governed by P3_FILE_FLAG, which directs Primer3 to generate files containing all acceptable primers evaluated. These files were used by many bioinformatics scripts that further evaluated the lists of primers and then selected pairs according to the needs of the particular application. Primer3 can now provide these primer lists on the output stream in boulder-IO format. Thus, applications that use primer3_core to generate these lists no longer need to manage temporary files.
Figure 8.

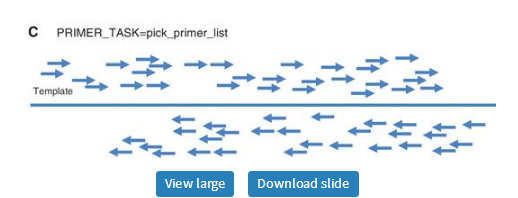
Three types of Primer3 task. Arrows represent PCR primers with 3′-ends at the arrow heads. Thick solid lines represent the template sequence. (A) The task generic directs Primer3 to design primer pairs for PCR when both forward (left) and reverse (right) primers are requested. Thin solid lines represent PCR products. (B) The task pick_sequencing_primers directs Primer3 to pick multiple unpaired primers for Sanger sequencing reads that will cover the template. Thin dashed lines represent Sanger sequencing reads. (C) The task pick_primer_list directs Primer3 to generate lists of legal forward and reverse primers without regard to their positions relative to each other.
The Primer3 manual (http://primer3.sourceforge.net/primer3_manual.htm) describes a few additional, more specialized, tasks.
Primer3 now offers a C/C++ programming library, named libprimer3, that offers the same primer-design functionality as the command line interface (Figure 1). The rationale is that some third-party programs using Primer3 functionality will benefit from the close integration offered by a library function-call interface. For example, the NCBI Primer-BLAST service (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (11) powers part of their primer design service with libprimer3.
Primer3 has two web interfaces—the original web interface, Primer3web, and the newer Primer3Plus (22), which is designed to be an easier-to-use, task-oriented interface that still harnesses the full power of Primer3. Primer3Plus offers several tabs from which all Primer3 parameters are accessible, including the new parameters controlling the thermodynamic models for secondary structure and those providing precise control over possible primer locations (Figure 2). We have set the parameters in Primer3Plus to reasonable default values, which should allow occasional users to design working primers by simply providing a template DNA sequence in the first tab of the web interface and clicking the ‘pick primers’ button. Experienced users can adjust arguments presented through the remaining tabs to exert fine control over the primer selection process.
When the user clicks the ‘pick primer’ button, the web application sends the template sequence and design parameters to primer3_core for primer selection (Figure 1). Primer3_core selects primer pairs and returns the boulder IO-formatted results to Primer3Plus, which parses these results and presents them to the user in graphical format (Figure 3). Users can then optionally store the primers using Primer3Manager, a simple tool for managing primer collections. We have updated Primer3Manager to support the extensible markup language (XML) based Real-time PCR Data Markup Language (RDML) format for primer information storage and exchange (25).
During the implementation of some of the new features described in this article, it became obvious that the primer search algorithm needed to be re-designed. The original algorithm looked for the best primer pairs by considering legal combinations of reverse and forward primers. It used a branch and bound approach based on the penalties associated with the individual forward and reverse primers. However, the algorithm inefficiently reevaluated possible pairs every time it had to find the next best pair. In the new implementation, to speed up the search, each pair is evaluated at most once, and then its characteristics are stored in memory. This increase in computational efficiency comes at the expense of higher memory usage. To minimize the amount of memory used, the new algorithm stores characteristics of primer pairs in hash maps and, therefore, allocates memory for a pair only when necessary. The algorithm also postpones computing some expensive-to-compute characteristics of primers until it is known that the primer might be part of a legal primer pair. These characteristics include prediction of the primer’s propensity to form hairpins or hybridize to other oligos or ectopic sites in the template and the primer’s propensity to amplify known high-abundance genomic repeats.
Over the last decade, Primer3 has proven to be a robust, versatile and widely used tool for primer selection. Evidence supporting these assertions stems both from the abundant use of Primer3 directly and from the many software programs and web services that rely on Primer3. The changing landscapes of molecular biology, genetics, genomics and bioinformatics have changed Primer3’s uses. These changes, plus experience with continued use of the program, indicated several opportunities for improvement of Primer3. We have augmented Primer3’s functionality and usability to take advantage of these opportunities. New capabilities include updated thermodynamic models for predicting oligo melting temperatures and thermodynamic models for predicting the propensity of oligos to form hairpin structures, to hybridize to each other or to hybridize to undesirable sites in template sequences. The new capabilities also include the ability to more precisely control the locations of primers. Additional enhancements improve ease of use and of integration with other software. These include the ability to require that primers not be used in multiple primer pairs, the ability to save and re-use argument settings, rationalization of the names of the boulder-IO arguments to primer3_core and a C/C++ programming library interface for calling Primer3 functionality directly from other programs. We are confident that, with the updates and improvements of the last several years, Primer3 and its web interfaces will remain useful and will serve as starting points for future primer design applications.
The Singapore Ministry of Health (to S.G.R.); the Singapore Agency for Science, Technology, and Research (to S.G.R.); the E.U. European Regional Development Fund through the Estonian Centre of Excellence in Genomics (to M.R. and T.K.); the Estonian Ministry of Education and Research [SF0180026s09 to M.R. and T.K]; National Science Foundation of the U.S.A. [DEB-1136626 to B.C.F.]; Intramural Research Program of the NIH, National Library of Medicine (to J.Y.). Funding for open access charge: The Singapore Ministry of Health (to S.G.R.); the Singapore Agency for Science, Technology, and Research (to S.G.R.).
Conflict of interest statement. None declared.