

发布者:primerbank 时间:2017-09-21 浏览量:966
William A. Walters, J. Gregory Caporaso, Christian L. Lauber, Donna Berg-Lyons,Noah Fierer, Rob Knight
Bioinformatics, Volume 27, Issue 8, 15 April 2011, Pages 1159–1161,https://doi.org/10.1093/bioinformatics/btr087
Published:23 February 2011

Using next-generation sequencing methods to characterize hundreds of samples simultaneously in a single sequencing run has revolutionized microbial ecology (Tringe and Hugenholtz, 2008). However, primer design for such studies remains challenging. The primers must amplify an appropriate region of DNA that is the right length for sequencing and also taxonomically informative (Liu et al., 2008; Wang et al., 2007); a linker that is not complementary to the target in any one of many diverse species must be inserted before the barcode to avoid differential amplification (Hamady et al., 2008); and the set of barcodes must be checked to avoid formation of secondary structure within or between primers (i.e. primer-dimers) or between the barcodes and the primers. Additionally, the techniques need to be generic rather than tied to one taxonomic outline or database, so that many different target genes can be studied.
Here we present PrimerProspector, an open-source software package for primer design and analysis built using the PyCogent toolkit (Knight et al., 2007), that resolves these issues. We recently applied PrimerProspector to identify the 16S rRNA 515f/806r primer pair as nearly universal to archaea and bacteria, and to optimize this primer pair for increased sensitivity across these domains. This optimized primer pair, applied successfully in several recent studies (Bates et al., 2010; Caporaso et al., 2010; G.Bergmann et al., manuscript in preparation), has provided novel insight into archaeal and bacterial community membership in soils by allowing for more accurate determination of the abundances of taxa missed by many commonly used canonical primer pairs, e.g. the Verrucomicrobia.
No existing tools specifically address the issues associated with designing barcoded polymerase chain reaction (PCR) primers for community analysis. Primer design is a large field and we cannot survey it comprehensively in this article, but among a selection of related tools, Primer Validator (http://bioinfo.unice.fr/454) allows taxonomic assessment but does not generate de novo primers, or allow a customizable 3′ weighted scoring system to predict successful amplification of tested primers. BarCrawl (Frank, 2009) allows design of barcodes for specified PCR primers but not design of the primers themselves, so is a useful complement to PrimerProspector. RDP's Probe Match (Cole et al., 2005) will report sequences matching a probe, as does Greengenes' probe function (DeSantis et al., 2006), but these tools are tied to the respective 16S rRNA databases and do not have support for barcodes. Primrose and OligoCheck (Ashelford et al., 2002) are useful for small numbers of target sequences, but do not scale well to thousands or tens of thousands of sequences, as is necessary when designing universal or near-universal primers, and do not incorporate differential weighting of 5′ and 3′ bases in primer scoring. Primer BLAST uses Primer3 software to build primers of a specified length against one target sequence, and then BLASTs the results against other databases to ensure that putative primers do not target BLAST hits. This functionality is also a useful complement to that provided in PrimerProspector.
While applications of PrimerProspector to date have focused on SSU rRNA primer design, PrimerProspector can be used for any nucleic acid sequences and allows users to design de novo primers based upon arbitrary multiple sequence alignments. User-specifiable design parameters include primer length, degeneracy and targeted regions for generation of primers. Existing or de novo primers can be analyzed for predicted taxonomic coverage, as shown in Figure 1. Finally, common pitfalls in primer design can be identified, such as likely barcode-primer secondary structure, regions susceptible to primer dimerization and disparate GC content between primer pairs. Convenient reports show amplicons or simulated reads that cover regions of sequences that are not phylogenetically informative or are of unsuitable lengths for sequencing.
Fig. 1.
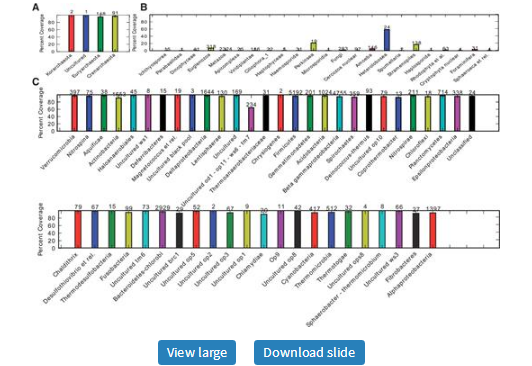
Taxonomic coverage summary of the 515f/806r 16S SSU rRNA primer pair at the phylum level for (A) archaea, (B) eukarya and (C) bacteria. The y-axes represent percent coverage and the value on top of each bar is the total number of reference sequences in each taxon. In this analysis, the reference sequences were derived from the Silva database, and filtered at 97% sequence identity with uclust (Edgar, 2010). Archaeal and bacterial sequences shorter than 1450 bases, and eukaryotic sequences less that 1800 bases, were excluded from the reference set. As illustrated, this primer pair is nearly universal for archaeal and bacterial 16S but is generally poor for eukaryotic (notably metazoan) 18S sequences. This plot and additional PrimerProspector analyses informed the decision to use this primer pair in Caporaso et al. (2010), Bates et al. (2010) and G.Bergmann et al. (2010). Comparisons with the unoptimized primer pair and with an alternative popular pair (27f/338r) are shown as Supplementary Figures S1 and S2, respectively.
De novo design of primers is performed by finding short conserved sequences in a given multiple sequence alignment to act as a 3′ binding site for new primers. Once these sites have been identified, full-length forward or reverse de novo primers are generated by incorporating the N upstream or downstream bases, where N is 15 by default. De novo full-length primers can then be sorted according to sensitivity, specificity or degeneracy, and compared with known primers to find matches or significant overlap. Specificity for particular target groups, such as archaea, can be obtained by supplying an optional alignment of sequences from which to exclude matches.
Primer analyses, including the prediction of taxonomic coverage, rely upon scoring primers against target sequences. To predict its taxonomic coverage, a primer is locally aligned to full-length target sequences with known taxonomies, and scored based on gap, 3′ mismatch and non-3′ mismatch counts. An example of the graphical output is provided in Supplementary Figure S3. The final five bases are considered to be the 3′ region by default, and are considered to be the most important for PCR amplification. The scoring scheme is parameterizable. The RDP Classifier (Wang et al., 2007) is used to classify the resulting sequence fragments, and the accuracy is displayed both in terms of which taxa are amplified and in terms of classification level of the resulting fragments. PrimerProspector supports retraining of the RDP Classifier for taxa coverage analysis based on different reference taxonomies.
Descriptions of the scripts included in PrimerProspector, the various outputs generated by PrimerProspector and an example based on the F515/R806 primer pair are included in the online documentation at http://pprospector.sourceforge.net/.
PCR amplification continues to be a key step in many high-throughput sequencing applications such as barcoded marker gene-based microbial community analyses. PrimerProspector represents a significant advance over prior work in this area by providing a single tool to facilitate primer design and analysis, including support for barcodes (and associated linkers). PrimerProspector is a fast and extensible framework for primer design and analysis, and has already been successfully applied to help researchers identify the most relevant and useful primers for their application, starting with multiple sequence alignments for any nucleic acid sequence.
Funding: Bill and Melinda Gates foundation; Crohn's and Colitis foundation of America; Howard Hughes Medical Institute; National Institutes of Health Signaling and Cell Cycle Regulation Training Grant (T32GM008759) in part.