

发布者:primerbank 时间:2017-09-21 浏览量:990
The taxonomic composition of a microbial community can be deduced by analyzing its rRNA gene content by, e.g., high-throughput DNA sequencing or DNA chips. Such methods typically are based on PCR amplification of rRNA gene sequences using broad-taxonomic-range PCR primers. In these analyses, the use of optimal primers is crucial for achieving an unbiased representation of community composition. Here, we present the computer program DegePrime that, for each position of a multiple sequence alignment, finds a degenerate oligomer of as high coverage as possible and outputs its coverage among taxonomic divisions. We show that our novel heuristic, which we call weighted randomized combination, performs better than previously described algorithms for solving the maximum coverage degenerate primer design problem. We previously used DegePrime to design a broad-taxonomic-range primer pair that targets the bacterial V3-V4 region (341F-805R) (D. P. Herlemann, M. Labrenz, K. Jurgens, S. Bertilsson, J. J. Waniek, and A. F. Andersson, ISME J. 5:1571–1579, 2011, http://dx.doi.org/10.1038/ismej.2011.41), and here we use the program to significantly increase the coverage of a primer pair (515F-806R) widely used for Illumina-based surveys of bacterial and archaeal diversity. By comparison with shotgun metagenomics, we show that the primers give an accurate representation of microbial diversity in natural samples.
PCR amplification and sequencing of 16S rRNA gene sequences directly from the environment has revolutionized our understanding of microbial diversity (1), in part because a significant fraction of microbes are difficult to grow in the laboratory (2). Due to development of next-generation sequencing technologies (3), we are experiencing another revolution in microbial ecology, since such studies now can be undertaken almost unconstrained by sequencing depth and number of samples (4–7). While shotgun metagenomics is growing in popularity for taxonomic profiling of samples, amplicon sequencing of highly informative taxonomic markers, such as the rRNA gene, is still considerably cheaper. However, approaches relying on PCR can alter the representation of taxa by amplification biases and, perhaps more so, by primer binding discrimination (8).
The coverage of a primer, i.e., the proportion of sequences within a given sequence set that is matched, can be improved by introducing degeneracies, meaning that alternative bases are used at one or more positions during the synthesis. The degeneracy of a sequence (termed d) is the number of unique sequence combinations it represents. Hence, the primer A(C/T)A(A/T/G)C has degeneracy, d, of 1 × 2 × 1 × 3× 1 = 6. While a higher degeneracy facilitates higher coverage, it also can lead to unspecific amplification. Therefore, degenerate primer design is a trade-off between specificity and coverage (sensitivity). In maximum coverage degenerate primer design (MC-DPD), the goal is to find a primer of length l, and maximum degeneracy, dmax, that matches a maximum number of sequences of a given input set, each of length l. Since the MC-DPD problem is NP complete (9) (i.e., an exact solution cannot be found in polynomial time), it needs to be addressed by using approximation heuristics.
The program HYDEN addresses the MC-DPD problem and was first used to design degenerate primers for a set of human genomic sequences in order to find new olfactory receptor genes (9). HYDEN uses an algorithm called Expansion, which for each window of length l within a given multiple sequence alignment tries to find the sequence of length l of highest coverage. It starts by finding the most frequent nucleotide at each position in the window and then combines these nucleotides into a sequence of d = 1. Subsequently, among the remaining nucleotides at all positions, it finds the nucleotide and position that has the highest frequency and adds this to the sequence. It repeats this procedure until d = dmax. It also uses the reverse approach, Restriction, going from full degeneracy at all positions and then removing nucleotides at different positions, in order of their increasing frequency, until d has dropped to dmax. This would be a good approach if no genetic linkage occurred between positions. However, this is often not the case, particularly not in structural RNA genes, such as rRNA. As an example, when designing a degenerate primer of dmax = 4 for the three input sequences AA, TT, and CC, an optimal primer would be, e.g., (A/T)(A/T), matching two out of three sequences. However, Expansion or Constriction would be equally likely to output the sequence A(A/T/C/G), only matching one sequence.
More recently, the program PrimerProspector was developed for design and evaluation of degenerate primers for taxonomic surveys (10). In a given multiple-sequence alignment, this program finds short (default of 5 bp) conserved sequences that will act as 3′ binding sites of primers. These will then be extended to form potential full-length primers. Degeneracies are allowed at every position in the primers, and the user can specify the minimal representation of a nucleotide in a position to be considered. Depending on the frequency distribution of nucleotides within the primer region, this will result in widely different degeneracy of the resulting primers. For instance, a minimal nucleotide representation of 40% in an 18-bp-long primer can give a d of 1 to 262,144. Hence, PrimerProspector does not address the MC-DPD problem.
Here, we present the program DegePrime, which is based on an algorithm we call weighted randomized combination for an approximate solution to the MC-DPD problem that preserves the correlation structure among nucleotides. We show that the program often outputs degenerate primers of higher coverage than HYDEN. In addition, the program can design primers based on over a million sequences, while HYDEN is limited to 2,000. DegePrime outputs the results in tabular format with the degenerate oligomer of dmax with highest coverage for every position of the sequence alignment, making it easy to select candidate primer combinations. It can also output coverage within different taxonomic groups of sequences. We used DegePrime earlier to design a primer pair, 341F-805R, for amplification of the V3-V4 region, which has been used in 454-based studies on a range of environments (11–16). This primer pair was shown to be the least biased among 512 primer pairs evaluated in silico for bacterial amplification and was experimentally shown to give a taxonomic composition similar to that of shotgun metagenomics (17). Here, we have used DegePrime to substantially improve the taxonomic coverage of a popular primer pair for amplification of the V4 region of bacterial and archaeal 16S. Finally, we compare the taxonomic composition that we obtain using our primer pairs in amplicon sequencing to that obtained with shotgun metagenomics on microbial communities from moose rumen and seawater.
Algorithm.For each window of an alignment, the algorithm tries to find a primer of length, l, and degeneracy, d ≤ dmax, matching as many sequences within the window as possible. A simplified version of the algorithm works as follows. First, the number of counts for each unique string of l in the window is counted. These strings then are combined in order of their frequency, starting with the most frequent string and adding one string at a time until the d of the combined sequence equals dmax. If (when d < dmax) the addition of a new string gives d> dmax, the algorithm instead tries adding the next string in frequency order until no more strings exist.
This will not always generate the optimal combination of strings. As an example, when running the algorithm with a dmax of 4 and an l of 2 on a window with five unique string sequences (CC, n = 30; AA, n = 20; GG, n = 10; CG, n = 10; GC, n = 10), the algorithm would output the degenerate sequence (A/C)(A/C), since CC followed by AA are the two most frequent strings. This matches 50 of the strings, while a combination of CC and GG into (C/G)(C/G) matched 60.
Hence, while it is generally a good idea to include strings of high counts, simply adding the strings in order of counts is not always best. As an alternative, random strings could be selected and combined, but when the number of unique strings is large, the probability of finding good combinations by chance is small. Instead, we use a combination of these two approaches. We select random strings among the observed strings but select them with probabilities proportional to their frequencies. New strings are selected this way until d = dmax. The coverage of the resulting primer is recorded, and the whole procedure is repeated 100 times. The best primer found this way for each window is output together with statistics on its coverage. We call this approach weighted randomized combination.
DegePrime software.DegePrime uses weighted randomized combination to find the degenerate primer with highest coverage for every window of a sequence alignment. When running DegePrime, the user specifies the parameters dmax and l. If dmax is not a possible degeneracy (these can be expressed as 2i × 3j, where i and j are integers or 0), it is automatically changed to the nearest lower possible degeneracy.
Since sequence alignments can include many gaps, the alignment optionally can first be processed to remove columns scarce in data using the script TrimAlignment. The user can either specify the minimum proportion of sequences that should have a nucleotide at an alignment column for this column to be kept in the processed file or refer to a reference sequence in the alignment, in which case the processed file will include the columns where the reference sequence has nucleotides. In order to provide DegePrime with information on where nucleotides have been deleted from sequences, which has implications for primer coverage calculations, the nucleotide upstream of a deleted nucleotide will be represented by a lowercase letter, while all other nucleotides will be represented by uppercase letters.
DegePrime can output the coverage of each primer among different groups of sequences, in which case an annotation file specifying the group label for each sequence is used as additional input to the program. The script MakeRdpTaxonomy can generate this annotation file from a GenBank file from the Ribosomal Database Project (RDP; http://rdp.cme.msu.edu) (18), and MakeSilvaTaxonomy can generate it from a fasta file from Silva (http://www.arb-silva.de) (19).
DegePrime, TrimAlignment, MakeRdpTaxonomy, and MakeSilvaTaxonomy were written in Perl. The software and source code for DegePrime are freely available at https://github.com/EnvGen/DegePrime.
Comparison with HYDEN.To enable comparison with HYDEN, a Perl script was written that instructs HYDEN to design a degenerate primer for every window of a sequence alignment.
Experimental procedures.One marine water sample and one moose (Alces alces) rumen sample were used to prepare shotgun sequencing libraries and 16S amplicon libraries using the PCR primer pairs 341F-805R and 515′F-805R, designed with DegePrime. The marine water sample was collected in the Kalmar Straight, Baltic Sea, and was captured on a 0.22-μm filter after removing larger particles by prefiltration through a 3.0-μm filter. DNA was extracted by phenol-chloroform and a proteinase K and lysozyme treatment as described in Riemann et al. (20) and further purified by ethanol precipitation. The moose rumen sample was collected from an animal in Småland, Sweden, within an hour after the animal was shot. DNA was extracted as described by Roume et al. (21). The extracted moose rumen DNA was submitted to the Science for Life Laboratory for TrueSeq library preparation and sequencing on an Illumina HiSeq (Illumina, Inc.), generating paired-end sequence reads that were 100 bp in length. The marine sample shotgun library was prepared in-house using Nextera according to the instructions of the manufacturer (Illumina, Inc., San Diego, CA, USA) and sequenced on an Illumina MiSeq (Illumina, Inc.), generating paired-end sequence reads that were 250 bp in length.
For the amplicon libraries, two consecutive PCR procedures were performed. The first one is aimed at amplifying the region of interest in the 16S gene, as well as attaching adapters to the amplicons that are used in the next step. For this, we used primers 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-NNNN-fwd_primer-3′ and 5′-AGACGTGTGCTCTTCCGATCT-rev_primer-3′, where NNNN are 4 random nucleotides that improve cluster definitions during sequencing, fwd_primer is either 341F (CCTACGGGNGGCWGCAG) or 515′F (GTGBCAGCMGCCGCGGTAA), as stated, and rev_primer is, in all cases, 805R (GGACTACHVGGGTWTCTAAT). The reaction mixtures were set up using 25 μl of Kapa HiFi master mix (Kapa Biosystems, Woburn, MA, USA), 2.5 μl of each primer (10 μM), 2.5 μl of template DNA (1 ng/μl), and 17.5 μl of water. These mixtures were submitted to thermocycling in a Mastercycler Pro S (Eppendorf, Hamburg, Germany) under the following conditions: 95°C for 5 min, 98°C for 1 min, 20 cycles of 98°C for 20 s, 51°C for 20 s, and 72°C for 12 s, followed by a final elongation step of 72°C for 1 min. Gel electrophoreses (1% agarose in 1× Tris-acetate-EDTA buffer) were carried out to check the size and quality of PCR products. Reaction products then were cleaned with magnetic beads and 15% polyethylene glycol 6000 in 1.5 M NaCl as described by Lundin et al. (22). Cleaning reduced the product volume to 23 μl in Tris-EDTA buffer, to which we added 25 μl of Kapa HiFi master mix and 1 μl of each of the primers 5′-AATGATACGGCGACCACCGAGATCTACACX8ACACTCTTTCCCTACACGACG-3 and 5′-CAAGCAGAAGACGGCATACGAGATX8GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′, where X8 is an Illumina-compatible barcode, such as the ones in the Nextera kit. In this way, each sequence can be uniquely identified during sequencing for a total of up to 96 samples using only 20 unique outer primers. These mixtures were subjected to 95°C for 5 min, 98°C for 1 min, 10 cycles of 98°C for 10 s, 62°C for 30 s, and 72°C for 15 s, followed by a final amplification step of 1 min on the same thermocycler as that described above and cleaned again using the same procedure. They then were delivered to Science for Life Laboratory/NGI (Solna, Sweden) to be sequenced on a MiSeq (Illumina, Inc., San Diego, CA, USA). For more detailed and updated protocols, see https://github.com/EnvGen/LabProtocols.
Sequencing data analysis.Amplicon sequences were quality trimmed using Fastx (http://hannonlab.cshl.edu/fastx_toolkit/links.html), trimming off bases from the 3′ end with a Phred score below 30. For clustering, all reads were trimmed to 220 bp; read pairs with one or both reads shorter than this were excluded. Forward and reverse reads then were concatenated. All samples were pooled and clustered at increasing similarities of 100%, 99%, and 98% using Usearch (23), keeping track of the read count coming from each sample. Representative sequences from each of the 98% similarity operational taxonomic units (OTUs) were converted back to separate 220-bp forward and reverse reads and run through the RDP classifier (24) for taxonomic assignments. The taxonomy generated by the RDP classifier was trimmed to keep, for each read, only taxonomic levels with at least 80% bootstrap value. If forward and reverse reads disagreed, we kept the longest consensus classification. If both reads agreed but one read of the pair was not classified as deeply as the other, it likely was because the one with the longest classification came from a more informative region of the 16S rRNA gene. Therefore, in this situation, the longest classification was kept. Finally, counts of taxonomic assignments were multiplied by the number reads for the OTU. Scripts for concatenating and splitting reads are available at https://github.com/EnvGen/Tutorials.
Shotgun reads, or partial reads, encoding 16S rRNA were extracted from the metagenome data using SortMeRNA (25) and were taxonomically classified with the RDP classifier using a cutoff of 80% bootstrap support. In cases where only one read in a pair had been extracted by SortMeRNA, its taxonomy (for as long as it had more than 80% bootstrap support) was used. If both reads in a pair had been extracted, the taxonomy for the pair was determined in the same way as that for the amplicon read pair data described above.
Comparison with HYDEN.We compared DegePrime and HYDEN in their ability to solve the maximum coverage degenerate primer design problem on aligned bacterial 16S rRNA gene sequences downloaded from RDP, v.9 (18). Since it is not possible to directly control the degeneracy of the primer with PrimerProspector, this program was not included in the comparison. HYDEN has a limit of 2,000 sequences, so we randomly subsampled 2,000 sequences from the larger file of 138,807 sequences and ran the comparison on this subset. Since the alignment contained many gaps, we first ran the script TrimAlignment to remove positions not represented in at least 90% of the sequences. We compared the coverage of the selected primers generated by the two programs at each alignment position with maximum degeneracy, dmax, set to 8, 24, and 128 and primer length, l, set to 18 (Fig. 1 shows results for dmax = 8). The two programs often suggested primers of the same coverage, but DegePrime generated higher coverage in 580, 602, and 539 positions at a dmax of 8, 24, and 128, respectively, while HYDEN generated primers of higher coverage in only 48, 119, and 207 positions for the same dmax settings. While DegePrime suggested primers of significantly higher coverage (P < 10−15 by Wilcoxon signed-rank test for every degeneracy level tested), the differences between the programs mostly occurred in regions of low conservation (Fig. 1). Both DegePrime and HYDEN outperformed a simplistic approach where a nondegenerate (d = 1) primer of maximum coverage was selected at each position (Fig. 1A). While HYDEN is restricted to 2,000 sequences, DegePrime can be run on much larger data sets; the processing of 1 million 16S rRNA gene sequences takes approximately 4 h on a MacBook Pro (Table 1).
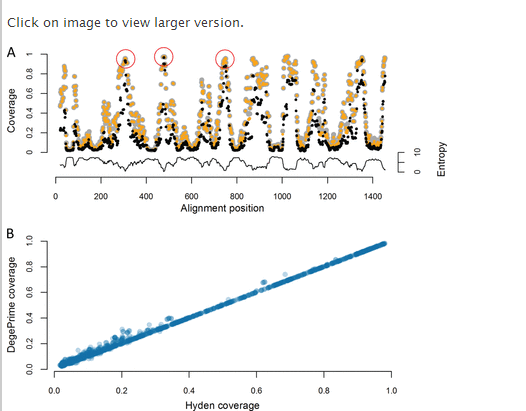

Design of broad-taxonomic-range PCR primers.We previously used DegePrime to design broad-range bacterial PCR primers suitable for the 454 Titanium sequencing platform with read lengths of 200 to 400 bp. The full data set described above, with 138,807 sequences, then was used to design primers amplifying the V3-V4 region of the bacterial 16S rRNA gene. High-coverage primers were found in the regions indicated by the red circles in Fig. 1, which correspond to Escherichia coli positions around 341 and 805. A set of primers with different lengths and degeneracies was tested with PCR and a primer pair 341F-805R [CCTACGGGNGGCWGCAG and GACTACHVGGGTATCTAATCC, with dvalues of 8 and 9, respectively; N is (A/G/C/T); W is (A/T); H is A/C/T; V is (A/C/G)] successfully amplified isolate and community DNA from different environments (Table 2 shows coverage among taxonomic groups). When run on biopsy samples rich in human cells, human DNA is sometimes amplified, but the ∼450-bp bacterial band can be separated from the ∼300-bp human band by excision from an agarose gel or by commercially available size-selective bead capture methods. The primer pair, supplemented with adapter and barcode sequences for multiplexing, has been successfully used in 454 sequencing applied to a wide range of environments (marine and lake water, lake sediments, and human gut samples [11–16]). In an evaluation of 512 primers by Klindworth et al. (17), this primer pair was found to give the least biased results for 454 sequencing of bacterial 16S rRNA genes.
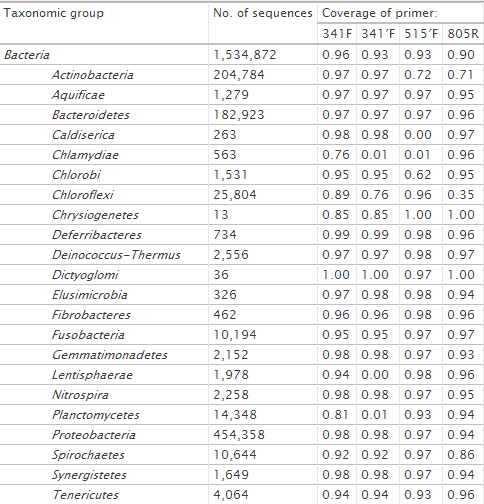
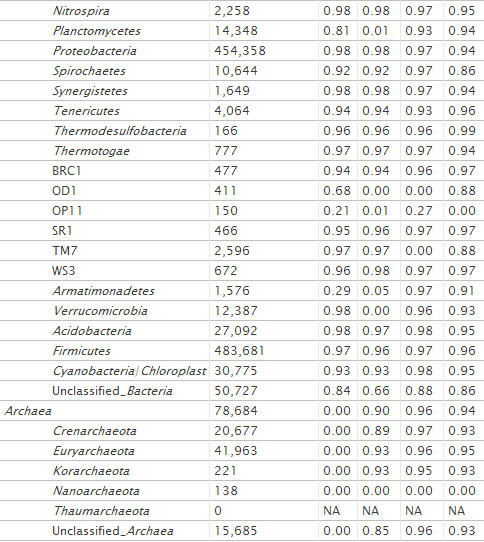

Primer 805R matches well to both archaea and bacteria (Table 2). However, 341F binds strictly to bacteria. Running DegePrime on a multiple alignment of archaeal sequences, we were able to identify positions where added degeneracy could render this primer capable of annealing to this domain of life as well. To limit the total degeneracy of the primer, we lowered the degeneracy at other positions. With a degeneracy of 18, this modified primer, 341′F [CCTAHGGGRBGCAGCAG; H is (A/C/T); R is (A/G); B is (C/G/T)], matches 93% of bacterial sequences and 90% of archaeal sequences. This level of degeneracy may require optimization of experimental conditions to avoid nonspecific amplification, especially in host-associated communities. Also, the primer misses some phyla that 341F matches well, like Chlamydiae, Lentisphaerae, Planctomycetes, and Verrucomicrobia (Table 2).
A primer pair, 515F-806R [GTGCCAGCMGCCGCGGTAA and GGACTACHVGGGTWTCTAAT, respectively; M is (A/C); H is (A/C/T); V is (A/C/G); W is (A/T)], amplifying the V4 region of the 16S rRNA gene, recently has been used successfully for Illumina sequencing (26) and is the primer pair used in the Earth Microbiome Project (www.earthmicrobiome.org). An attractive feature of this primer pair is that it should match bacteria as well as archaea (www.earthmicrobiome.org). However, a test using the Probe Match tool in RDP (18) reveals that the forward primer matches only 53% of archaea; it misses nearly all crenarchaea and unclassified archaea (see Table S1 in the supplemental material). Similar results were obtained with Greengenes Probe Locator (27 and data not shown). We ran DegePrime on 36,881 archaeal 16S sequences downloaded from RDP (v.10) with dmax = 2 (same degeneracy as the 515F primer described above) and l = 19. At the position of the 515F primer, DegePrime output the primer GTGYCAGCCGCCGCGGTAA [Y is (C/T)]; hence, it chose to use the degeneracy at a position other than that in the original primer. This primer matches 93% of archaeal sequences in RDP. When we increased the allowed degeneracy to 6, DegePrime suggests the primer GTGBCAGCMGCCGCGGTAA [B is (C/G/T); M is (A/C)], which covers 96% of archaeal and 93% of bacterial sequences. We call this primer 515′F; its taxonomic coverage is described in Table 2.
Experimental evaluation.To assess whether the primers designed with DegePrime give an unbiased view of community composition in natural microbial communities, shotgun libraries and amplicon libraries from primer pair 341F-805R and 515′F-805R were prepared from two samples: one marine surface water sample and one moose rumen sample. From the shotgun sequences, reads of 16S rRNA genes were extracted and taxonomically classified. From the amplicon libraries, reads were clustered to operational taxonomic units (OTUs) of 98% similarity before classification. On average, 99.8% of the amplicon reads were assigned to OTUs that could be classified as either bacterial or archaeal 16S (with 80% bootstrap support), showing that the primer pairs are specific to the 16S rRNA gene despite their degeneracies. For both samples, the counts of taxonomic groups correlated well between the shotgun and amplicon sequencing data (Fig. 2). All microbial classes detected with at least 1/1,000 reads in the metagenomes also were detected in the amplicon data sets, with the only exception being Spirochaetes, with 0.5% of reads in the moose rumen metagenome but undetected using the two primer pairs. Conversely, there was just a single class with >1/1,000 reads in one of the amplicon data sets that was undetected in the corresponding metagenome data set: the Planctomycetacia, with 0.3% reads in the moose rumen sample amplified with primer 515′F. The reasons for these discrepancies are not clear. All primers match ≥95% of Treponema sequences in RDP, which is the dominating Spirochetes genus in the rumen, but it may be that the dominant strains in the sample have mismatches relative to the primers. The seeming overamplification of Planctomycetacia with primer 341F in the moose rumen sample (but not in the water sample) is harder to explain but may be attributed to random noise.
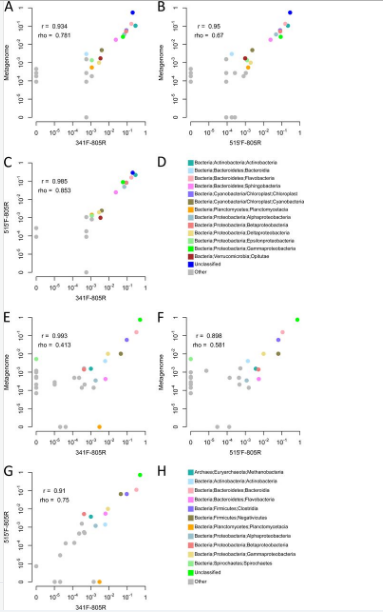

According to the metagenome data, both samples contain only small amounts of archaea, with 0.1% and 0.3% in the water and rumen sample, respectively. In accordance with its better matching to archaea, primer 515′F generates more archaeal sequences than 341F for both samples, 0.009% versus 0% for the water sample and 0.5% versus 0.1% for the rumen sample. For both samples, the shotgun data had a higher proportion of reads than the amplicon data that could not be classified to the class level using 80% bootstrap support. While this may reflect that rare taxa not yet included in the databases are picked up by shotgun sequencing to a greater extent than by amplicon sequencing, more likely it is a product of short shotgun reads obtained from uninformative regions of the 16S gene that do not carry enough information for taxonomic classification at this level. For the same reason, it is not possible to fairly compare the profiled communities at finer taxonomic levels.
We also used the shotgun metagenome data to evaluate how much coverage is gained by using our degenerate primers compared to using nondegenerate primers by using in silico matching of the primers to the 16S rRNA shotgun reads. On average, 94% of the reads matching the degenerate primer also matched the best nondegenerate primer. The difference was most pronounced for 805R, where for both samples only 88% of the reads matching the degenerate primer also matched the best nondegenerate primer (see Table S2 in the supplemental material).
Conclusions.We present the program DegePrime, which uses a new algorithm, which we call weighted randomized combination, for solving the maximum coverage degenerate primer design problem. We have demonstrated the utility of DegePrime for designing broad-taxonomic-range degenerate PCR primers. We show that amplicon libraries generated with the 16S primers proposed by DegePrime faithfully reconstruct the community profiles obtained with shotgun sequencing. We believe this program will be applicable for designing primers for other taxonomic markers and for gene families of medical or technological interest. To the best of our knowledge, there is no other tool currently available that can process such a large number of sequences while producing primers with size and maximum degeneracy specified by the user. Further improvements to this software could include predictions of hairpin formations and primer dimers and calculations of annealing temperatures that would aid in the final primer selection, as well as the option to specify reference sequences that primers are not allowed to match.